Introduction
Hepatocellular carcinoma (HCC) poses a significant
challenge to global health, being the third leading cause of
cancer-related mortality. Although early detection can lead to a
favorable prognosis through surgical resection, achieving a patient
survival rate of >70% (1), most
cases are diagnosed at advanced stages. In China, the 5-year
survival rate is <12.5% (2).
Therefore, safe and effective treatments are urgently needed.
In pursuit of innovative and targeted treatment
strategies, the integration of radioisotope-based therapies has
emerged as a promising field for precision medicine. Among these,
iodine-125 (125I) seed brachytherapy has been widely
used in the clinical treatment of various types of cancer (3). The combination of 125I
radioactive particle implantation with epidermal growth factor
receptor tyrosine kinase inhibitors (EGFR TKIs) has shown superior
efficacy and long-term survival in patients with advanced non-small
cell lung cancer compared with EGFR TKIs alone. In addition, this
combination therapy has been reported to regulate the expression of
T-lymphocyte subsets, natural killer cells and immune-inflammatory
factors, thereby improving immune function (4). Additionally, lobaplatin-transarterial
chemoembolization (TACE) combined with radioactive 125I
seed therapy has been shown to enhance disease control and overall
survival in patients with primary HCC (5). Furthermore, a combination therapy of
125I seed implantation with TACE can substantially
prolong the median survival time, and improve the 6-, 12- and
18-month survival rates of patients with HCC plus portal vein tumor
thrombosis (6). Despite these
advances, the precise mechanism of action of 125I
radiation particles in liver cancer treatment remains unclear.
Invasion and metastasis are the fundamental
characteristics of HCC (7).
Understanding the cellular signaling mechanisms driving cancer
transformation, as well as those governing cell proliferation,
invasion and angiogenesis, may provide valuable insights into
therapeutic mechanisms (8). It has
previously been indicated that 125I upregulates the
PERK-eIF2a-ATF4-CHOP pathway to promote apoptosis. Notably, the
ATF4-CHOP pathway is crucial in endoplasmic reticulum stress, and
induces cell apoptosis by upregulating CHOP, Bcl-2 and other
apoptosis-related factors (9). The
inhibition of glycolysis can also enhance the inhibitory effects of
radiotherapy on cancer cell proliferation, invasion and migration
(10,11). Notably, 125I has been
reported to inhibit glycolysis in HCC by regulating the
microRNA-338/PFKL axis, thus affecting the Warburg effect (12). However, the mechanism by which
125I inhibits tumor progression remains unclear.
In the present study, human liver cancer cells
(MHCC-97H) were subjected to intervention with 125I; the
cells were divided into a control group and an 125I
intervention group. The collected samples from both groups
underwent comprehensive transcriptomics and proteomics analyses to
elucidate changes in gene expression and functional alterations at
the protein level. Integration of these two sets of data allowed
for the identification of genes exhibiting notable differences in
both transcriptional and protein expression, characterization of
the biological functions of these differentially expressed genes,
and delineation of potential pathways associated with tumor
inhibition. This integrated approach not only provides a holistic
understanding of the molecular changes induced by 125I
intervention in liver cancer cells, but also offers novel insights
and a theoretical foundation for the mechanism underlying the
therapeutic effects of 125I in liver cancer
treatment.
Materials and methods
Cell culture
The MHCC-97H HCC cell line was purchased from
Cellverse Bioscience Technology Co., Ltd., and 125I was
procured from Shanghai Xinke Pharmaceutical Co., Ltd. The cells
were cultured in 90% Dulbecco's modified Eagle's medium (Beijing
Solarbio Science & Technology Co., Ltd.) supplemented with 10%
fetal bovine serum (FBS; Cyagen Biosciences, Inc.) and 1%
penicillin/streptomycin mixture (Beijing Solarbio Science &
Technology Co., Ltd.), which was used to maintain cell viability
and prevent contamination. The cells were cultured at 37°C and 5%
CO2. The cells were grouped as follows: Control group,
normal MHCC-97H cells; and 125I group. For
125I radiation of MHCC-97H cells, the initial activity
and dose rate were 3.0 mCi and 3.412 cGy/h, respectively. Cells
were irradiated with 125I at a dose of 0.82 Gy for 24 h
37°C (9). 125I seeds
were purchased from Shanghai Xinke Pharmaceutical Co., Ltd.
Cell Counting Kit-8 (CCK8) assay
To assess the impact of 125I intervention
on MHCC-97H cells, the CCK8 assay (US Everbright, Inc.) was
employed. MHCC-97H cells were seeded in 96-well plates at a density
of 2,000 cells/well and were treated with 125I.
Subsequently, the cells were incubated with 20 µl CCK8 reagent for
4 h and the cell viability was determined by measuring absorbance
at a wavelength of 450 nm. Each CCK8 assay was performed with five
biological replicates to ensure the reliability of the results.
5-Ethynyl-2′-deoxyuridine (EdU)
assay
The 5-ethynyl-2′-deoxyuridine (EdU) assay (US
Everbright, Inc.) was conducted to evaluate the effects of
125I intervention on MHCC-97H cells. When the cell
density reached 85–90%, MHCC-97H cells were incubated with 50 µM
EdU reagent for 2 h at room temperature and were subsequently fixed
using 4% paraformaldehyde for 0.5 h at room temperature. EdU
staining was performed using 1X Apollo (fluorescent dye) and DNA
was stained with 1X Hoechst 33342 solution at room temperature in
the dark for 0.5 h. The stained cells were subsequently visualized
under a fluorescence microscope. Each EdU assay was performed in
triplicate to ensure the robustness and reproducibility of the
results.
Cell colony formation assay
A single-cell suspension of MHCC-97H cells was
inoculated into a 6-well plate at a density of 500 cells/well. The
cells were incubated at 37°C for 12 days to facilitate colony
formation. Subsequently, the cells were rinsed twice with PBS,
fixed with 1 ml methanol at room temperature for 15 min and stained
with 0.3% crystal violet (Wuhan Servicebio Technology Co., Ltd.)
for 5 min at room temperature. Colonies were manually counted with
those containing >50 cells counted.
Wound healing assay
MHCC-97H cells were inoculated in 6-well plates, and
when the cell monolayer reached 90% confluence, a 200-µl pipette
tip was used to gently scratch the monolayer across the center of
the well. Subsequently, the cells were washed with PBS and cultured
in complete medium (1% FBS) (13)
for 24 h. Images of the scratches were captured at 0 and 24 h under
an inverted light microscope.
Transwell assay
The 24-well Transwell chambers (pore size, 8 µm)
were coated in 80 µl Matrigel (Matrigel:serum-free medium, 1:8;
cat. no. 356234; Corning, Inc.) in 37°C for 3 h. Subsequently,
5×104 MHCC-97H cells suspended in serum-free medium were
added to the upper compartments of Transwell chambers, whereas the
lower compartments were filled with culture medium containing 10%
FBS. After 48 h of cultivation, the cells remaining in the upper
compartments were removed with cotton swabs, and the cells that had
penetrated the membrane were stained with 0.3% crystal violet for
10 min at room temperature. The number of invasive cells was
counted manually using a light microscope. Transwell assays were
performed in triplicates.
Flow cytometry
The Annexin V-FITC Apoptosis Detection Kit (Biosharp
Life Sciences) was used to detect the level of apoptosis. MHCC-97H
cells (1×105) were collected in PBS and suspended in 100
µl binding buffer. Subsequently, 5 µl Annexin V-FITC was added to
the binding buffer and incubated with MHCC-97H cells for 10 min at
room temperature in the dark. PI (10 µl) was then added, gently
mixed, and incubated for 5 min at room temperature in the dark,
followed by the addition of 400 µl PBS to resuspend the cells. Cell
samples were loaded onto a flow cytometer (CytoFocus421 instrument;
Beijing Zhizhen Biological Technology Co., Ltd.) for detection.
Flow cytometry using the CytoFocus421 instrument and CytoFocus 3.2
software (Beijing Zhizhen Biological Technology Co., Ltd.) was
performed in triplicate
Transcriptomics analysis
Preparation of transcriptome samples
For transcriptomic analysis, untreated cells served
as the control group, whereas cells treated with 125I
constituted the 125I intervention group. Three pairs of
samples were selected for the transcriptomics analysis. Initially,
total RNA was extracted from the cells using QIAzol lysis reagent
(Qiagen, Inc.), RNA concentration and purity were assessed using a
Nanodrop 2000 (Thermo Fisher Scientific, Inc.), and RNA integrity
was confirmed by agarose gel electrophoresis (1% agarose gel;
Biowest) using the SYBR™ Green I Nucleic Acid Gel Stain (Thermo
Fisher Scientific, Inc.). The RNA Integrity Number value was
measured using an Agilent 2100 Bioanalyzer (Agilent Technologies,
Inc.). For single library construction, a total RNA volume of ≥1 µg
and a concentration of ≥35 ng/µl was required. The optical density
ratios OD260/280 ≥1.8 and OD260/230 ≥1.0 were used as indicators of
RNA purity. These stringent criteria were implemented to ensure
high quality and integrity of the RNA samples, thereby guaranteeing
the reliability of subsequent transcriptomics analyses.
Database construction and
sequencing
To construct the database and facilitate sequencing,
magnetic beads with Oligo (dT) and polyA (Thermo Fisher Scientific,
Inc.) were employed for A-T base pairing to selectively isolate
mRNA from total RNA samples. Paired-end sequencing was conducted
using an Illumina HiSeq 4000 platform (Illumina, Inc.) owing to its
advanced capabilities for high-throughput sequencing. The
sequencing length was 2×150 bp, the final library concentration was
1–20 pM and its concentration was measured using the Qubit ssDNA
Assay Kit (cat. no. Q10212; Thermo Fisher Scientific Inc.).
Sequencing was performed using the HiSeq 3000/4000 SBS Kit (cat.
no. FC-410-1003; Illumina, Inc.). Fragmentation buffer was
introduced to randomly break down the mRNA, ensuring the generation
of representative fragments for analysis. A small fragment of ~300
bp was selectively screened and isolated using magnetic beads,
ensuring the isolation of fragments of interest. The isolated
fragments were reverse transcribed to synthesize cDNA using the
High-Capacity cDNA Reverse Transcription Kit (cat. no. 4368814;
Thermo Fisher Scientific, Inc.) with the following steps: 25°C for
10 min, 37°C for 120 min and 85°C for 5 min, which is a crucial
step for subsequent sequencing analysis. The double-stranded cDNA
has sticky ends, and EndRepairMix (cat. no. Y9140; Qiagen, Inc.)
was added to convert them into blunt ends, followed by the addition
of an A base at the 3′ end to facilitate the subsequent ligation of
the adapter sequence.
Fragment screening and library enrichment,
purification and fragment sorting of the products connected to the
adapter, PCR amplification of the sorted products, and purification
were performed to obtain the final library.
Analysis of raw sequencing data
Analysis of raw sequencing data involved a series of
steps to ensure data quality and extract meaningful insights.
Transcriptomics analysis was conducted using the following
statistical methods: fastp (https://github.com/OpenGene/fastp) was employed to
evaluate and screen the quality of the raw sequencing data obtained
from Illumina sequencing, ensuring the reliability of subsequent
analyses. RSEM (http://deweylab.github.io/RSEM/) was applied for the
quantitative analysis of both chain-specific and non-chain-specific
transcriptomics data. This approach provides an estimate of
transcript abundance, contributing to an overall understanding of
gene expression levels. Transcripts per million was used to
standardize the gene expression levels. This normalization method
allowed the comparison of gene expression across different samples,
considering variations in library sizes. DESeq2 (http://bioconductor.org/packages/stats/bioc/DESeq2/),
a robust tool for RNA-Seq data analysis, was employed to identify
differentially expressed genes (DEGs) between the control and
125I intervention groups. DEGs were filtered based on
specific criteria, including expression difference multiples (|
log2FoldChange |) ≥1, a false discovery rate (FDR) <0.5 and
P<0.05. The differential gene functional enrichment analyses
included Kyoto Encyclopedia of Genes and Genomes (KEGG; Version
2022.10; http://www.genome.jp/kegg/); Gene
Ontology (GO) analysis, which includes Biological Process (BP),
Cellular Component (CC) and Molecular Function (MF) (goatools;
Version 0.6.5; http://pypi.org/project/goatools/); Reactome (Version
82; http://reactome.org); and Disease Ontology (DO;
http://disease-ontology.org) enrichment
analyses.
Proteomics analysis
Sample preparation
For proteomics analysis, three cell samples from
each group (control and 125I intervention groups) were
selected. The sample preparation process involved the following
steps: Total cell protein was extracted from the selected cell
samples using RIPA buffer (Thermo Fisher Scientific, Inc.) to
capture the complete proteomics profile. The bicinchoninic acid
method was employed for protein quantification to ensure accurate
measurement of protein concentrations in the samples. Subsequently,
100 µg protein sample was supplemented with lysis buffer to a final
volume of 90 µl. A final concentration of 10 mmol/l TCEP reducing
agent was added and the mixture was incubated at 37°C for 60 min. A
final concentration of 40 mmol/l iodoacetamide was then added and
was incubated in the dark at room temperature for 40 min. Precooled
acetone (ratio of acetone to sample volume, 6:1) was added to each
tube, followed by precipitation at −20°C for 4 h. After
centrifugation at 10,000 × g for 20 min at 4°C, the precipitate was
collected. The sample was fully dissolved in 50 mmol/l TEAB and
trypsin was added at a mass ratio of 1:50 (enzyme:protein) for
enzymatic digestion overnight at 37°C. TMT labeling and mixing were
performed; the TMT reagent (cat. no. 9011; Thermo Fisher
Scientific, Inc.) was brought to room temperature, followed by the
addition of acetonitrile and vortexing. For every 100 µg peptide,
one vial of TMT reagent was added (TMT10-126 for labeling). The
mixture was incubated at room temperature for 2 h, after which,
hydroxylamine was added and the reaction was carried out at room
temperature for 15 min. The labeled products were mixed together in
equal amounts in one tube and dried using a vacuum concentrator;
this step facilitated protein identification and quantification.
The peptide samples were solubilized in ultra-performance liquid
chromatography buffer to ensure that the samples were suitable for
subsequent analysis. A C18 column was used for high-pH liquid-phase
separation, which enabled the separation of peptides based on their
physicochemical properties.
Liquid chromatography-tandem mass
spectrometry (MS/MS)
Nanoscale liquid chromatography-MS/MS technology
(Easy-nLC1200 coupled with QExactive mass spectrometer; Thermo
Fisher Scientific, Inc.) was used in the present study. Ionization
mode, positive; nitrogen gas temperature, 350°C; nebulizer
pressure, 40 psi. Peptides were dissolved in mass spectrometry (MS)
loading buffer, and after loading, they were separated through a
C18 chromatography column (75 µm × 25 cm; Thermo Fisher Scientific,
Inc.) for 120 min at a flow rate of 300 µl/min. The EASY-nLC liquid
phase gradient elution was performed as follows: Phase A, 2%
acetonitrile with 0.1% formic acid; Phase B, 80% acetonitrile with
0.1% formic acid; 0–1 min, 0–5% B; 1–63 min, 5–23% B; 63–88 min,
23–48% B; 88–89 min, 48–100% B; 89–95 min, 100% B. The MS and MS/MS
acquisition switched automatically, with MS resolutions of 70 and
35K, respectively. MS was used to perform a full scan (m/z
350–1300), and the top 20 parent ions were selected for secondary
fragmentation with a dynamic exclusion time of 18 sec.
Data analysis
For data analysis, Proteome Discoverer Software 2.2
(Thermo Fisher Scientific, Inc.) was employed. Peptide
identification was controlled for accuracy by setting the FDR to
FDR ≤0.01, ensuring a reliable identification of peptides.
Student's unpaired t-test was used to calculate the P-value of
inter-sample differences and the fold change (FC) between groups.
This analysis aimed to identify proteins with significant changes
in expression in response to 125I treatment.
Significantly differentially expressed proteins were identified
based on specific criteria: Proteins with P<0.05 and FC >1.2
were considered upregulated, whereas those with P<0.05 and FC
<0.83 were considered downregulated. The differential protein
functional enrichment analyses included KEGG; and GO analysis of
BP, CC and MF. In addition, Evolutionary Genetics of Genes: Non
superior Orthologous Groups (EggNOG; version 2020.06; http://eggnogdb.embl.de/#/app/home)was used to
determine protein functional classification; and subcellular
localization prediction was performed using WoLF PSORT (https://wolfpsort.hgc.jp/), which determines the
location of proteins within cells.
Comprehensive analysis
By conducting a Venn joint analysis, the present
study screened differentially expressed genes from transcriptomic
and proteomic data. Subsequently, the STRING (https://string-db.org/) database was utilized to
perform protein-protein interaction network analysis. In addition,
the Xiantao (https://www.xiantao.love/products) software platform
was used for further bioinformatics analysis, in the analysis, the
samples were independent, with an equal number of adjacent normal
tissues and cancer tissues. However, these samples were not from
the same group of patients. Therefore, the Wilcoxon Rank Sum Test
was used for statistical analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, the RNA was subjected to phenol-chloroform extraction
for further purification. The quantity and quality of the purified
RNA were assessed by measuring the absorbance at 260/280 nm using a
microplate reader (Thermo Fisher Scientific, Inc.), and the
acceptable ratio for A260/A280 was considered 1.8–2.2.
Subsequently, cDNA was synthesized (PrimeScript RT reagent Kit;
Takara Bio, Inc.) using the following standard procedure: 37°C for
15 min and 85°C for 5 sec, followed by maintenance at 4°C. The qPCR
(PowerUp SYBR Green Master Mix; Thermo Fisher Scientific, Inc.)
procedure was as follows: 95°C for 1 min, followed by 40 cycles at
95°C for 10 sec and 60°C for 30 sec. Each transcript concentration
was normalized to the mRNA expression levels of GAPDH using the
2−ΔΔCq method (14).
The primer sequences were as follows: GAPDH, forward
5′GGTCGGAGTCAACGGATTTG-3′, reverse 5′-GGAAGATGGTGATGGGATTTC-3′;
GPNMB, forward 5′-CTTCTGCTTACATGAGGGAGC-3′, reverse
5′-GGCTGGTGAGTCACTGGTC-3′; C4BPA, forward
5′-ATGACCTTGATCGCTGCTCTG-3′, reverse 5′-GTCAACGTAATATCCATCGGGG-3′;
MT2A, forward 5′-TCCTGCAAATGCAAAGAGTGC-3′, reverse
5′-GTTTGTGGAAGTCGCGTTCT-3′; CTH, forward
5′-CATGAGTTGGTGAAGCGTCAG-3′, reverse, 5′-AGCTCTCGGCCAGAGTAAATA-3′;
and H1-0, forward 5′-ACTCGCAGATCAAGTTGTCCA-3′, reverse
5′-GGTTCGTCGCTCTTGGCTA-3′.
Statistical analysis
Data are presented in bar graphs, with each
experiment conducted independently three times. The data from the
experiments are presented as the mean ± standard error of the mean.
All data calculations and statistical analyses were carried out
using SPSS 23.0 (IBM Corp.) and GraphPad Prism 6.01 (Dotmatics).
For the comparison of two consecutive variables, the statistical
significance of normally distributed variables was analyzed using
unpaired Student's t-test, whereas the differences between
non-normally distributed variables were analyzed using Wilcoxon
rank-sum test. P<0.05 was considered to indicate a statistically
significant difference.
Results
125I intervention
suppresses MHCC-97H cell viability, proliferation, invasion and
migration, and induces cell apoptosis
The present study initially investigated the effect
of 125I on the viability and proliferation of MHCC-97H
cells (Fig. 1A-C). After 24 h of
125I treatment, cell viability was significantly reduced
(Fig. 1A) and the proportion of
EdU-positive cells was significantly decreased (Fig. 1B). Furthermore, the number of cell
colonies significantly decreased after 12 days of 125I
treatment (Fig. 1C). The present
study also examined the effects of 125I on the invasion
and migration of MHCC-97H cells (Fig.
1D and E). The wound healing area was significantly decreased
after 24 h of 125I treatment (Fig. 1D), and the number of invasive cells
was significantly decreased after 48 h of 125I
intervention (Fig. 1E). Flow
cytometry revealed that the percentage of PI-positive cells was
significantly increased after 24 h of 125I treatment
(Fig. 1F). These results indicated
that 125I intervention may inhibit MHCC-97H cell
proliferation, invasion and migration, and promote cell
apoptosis.
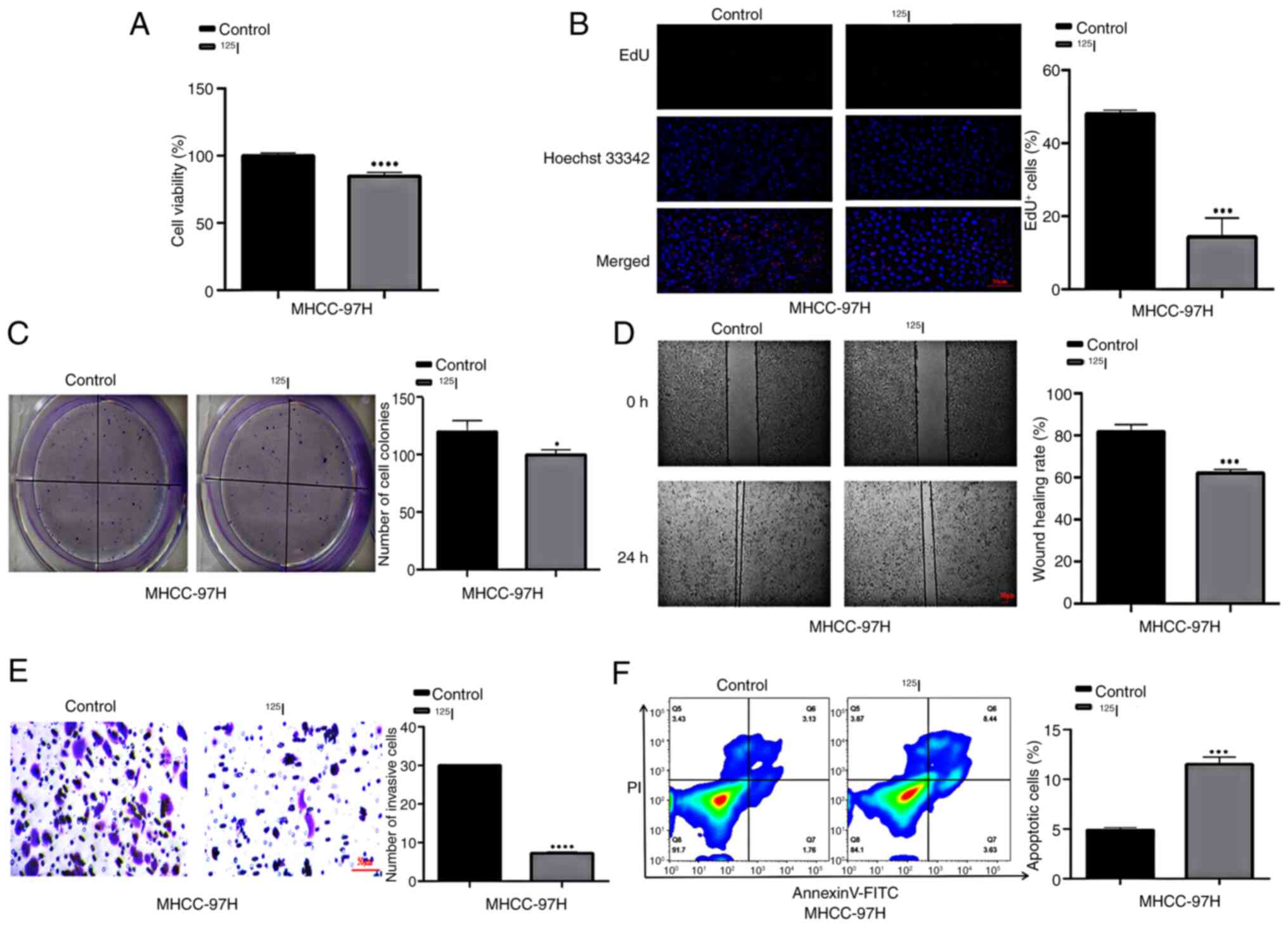 | Figure 1.125I suppresses HCC cell
viability, proliferation, invasion and migration, and induces
apoptosis. Cell viability and proliferation were assessed using (A)
Cell Counting Kit-8 assay, (B) EdU assay (scale bar, 50 µm) and (C)
colony formation assay. Cell invasion and migration assay were
assessed using (D) wound healing assay (scale bar, 50 µm) and (E)
Transwell assay (scale bar, 50 µm). (F) Flow cytometric analysis of
cell apoptosis and PI staining. *P<0.05, ***P<0.001,
****P<0.0001 vs. control. EdU, 5-ethynyl-2′-deoxyuridine;
125I, iodine-125. |
Transcriptomics analysis of
125I intervention in MHCC-97H cells
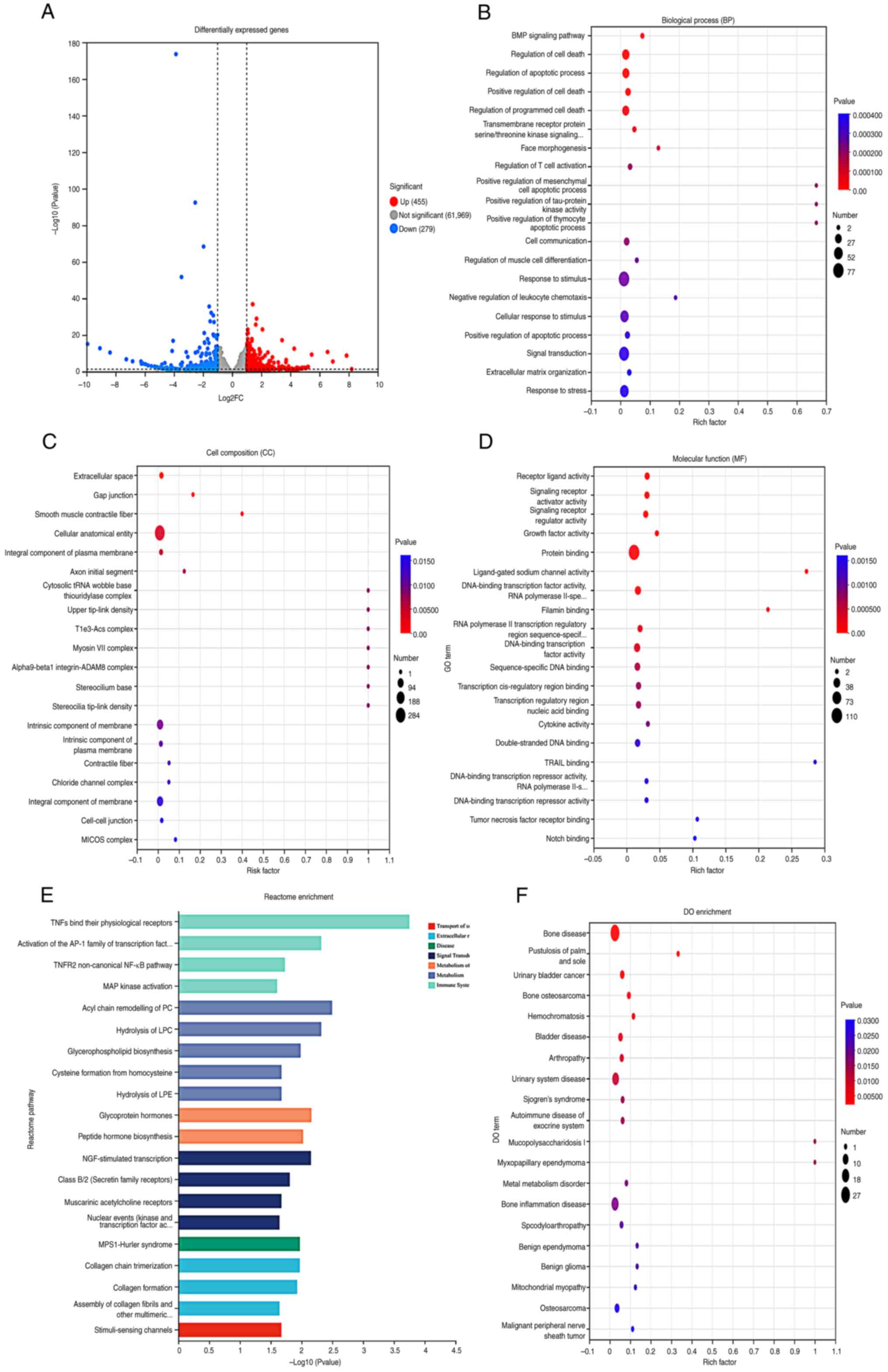
The differential expression analysis revealed that
there were 734 differentially expressed genes between the control
group and the 125I intervention group, with 455 genes
being upregulated and 279 genes being downregulated. These results
were visualized using a volcano plot, where the red dots represent
significantly upregulated proteins, blue dots represent
significantly downregulated proteins, and gray dots represent
proteins with no differential expression (Fig. 2A). Compared with in the control
group, the differentially expressed genes in the 125I
intervention group were primarily involved in ‘BMP signaling
pathway’, ‘Regulation of programmed cell death’ and ‘Regulation of
apoptotic process’ in BP terms (Fig.
2B). In terms of CC terms, the differentially expressed genes
were mainly enriched in ‘Extracellular space’, ‘Gap junction’ and
‘Smooth muscle contractile fiber’ (Fig. 2C). MF term analysis indicated that
the differentially expressed genes were predominantly enriched in
‘Receptor ligand activity’, ‘Signaling receptor activator activity’
and ‘Signaling receptor regulator activity’ (Fig. 2D). Reactome pathway enrichment
analysis revealed that the differentially expressed genes were
mainly enriched in processes such as ‘TNFs bind their physiological
receptors’, ‘Activation of the AP-1 family of transcription factor’
and ‘TNFR2 non-canonical NF-kB pathway’ (Fig. 2E). DO database analysis revealed
that the differentially expressed genes were related to diseases,
such as ‘Bone disease’, ‘Pustulosis of palm and sole’ and ‘Urinary
bladder cancer’ (Fig. 2F).
Proteomics analysis of 125I
intervention in MHCC-97H cells
Volcano plot analysis revealed significant
differences in the expression of 387 proteins between the control
and 125I intervention groups, with 277 upregulated and
110 downregulated proteins (Fig.
3A). Functional enrichment analysis of the proteins was
performed using Gene Ontology to reveal the BP, CC and MF terms in
which they were involved. Regarding BP terms, differentially
expressed proteins participated in responses to biological stimuli,
humoral immune responses, defense responses and reactions to
external biological stimuli (Fig.
3B). KEGG enrichment analysis showed that the differentially
expressed proteins were involved in ‘Complement and coagulation
cascades’, ‘Pertussis’ and ‘Metabolic pathways’ (Fig. 3C). The EggNOG classification
indicated that the differentially expressed proteins were
associated with processes such as post-translational modification,
protein turnover, molecular chaperones, intracellular transport,
secretion, vesicular transport and transcription (Fig. 3D). Subcellular localization
prediction (WoLF PSORT) suggested that the differentially expressed
proteins were primarily localized in the cytoplasm, nucleus and
endoplasmic reticulum (Fig.
3E).
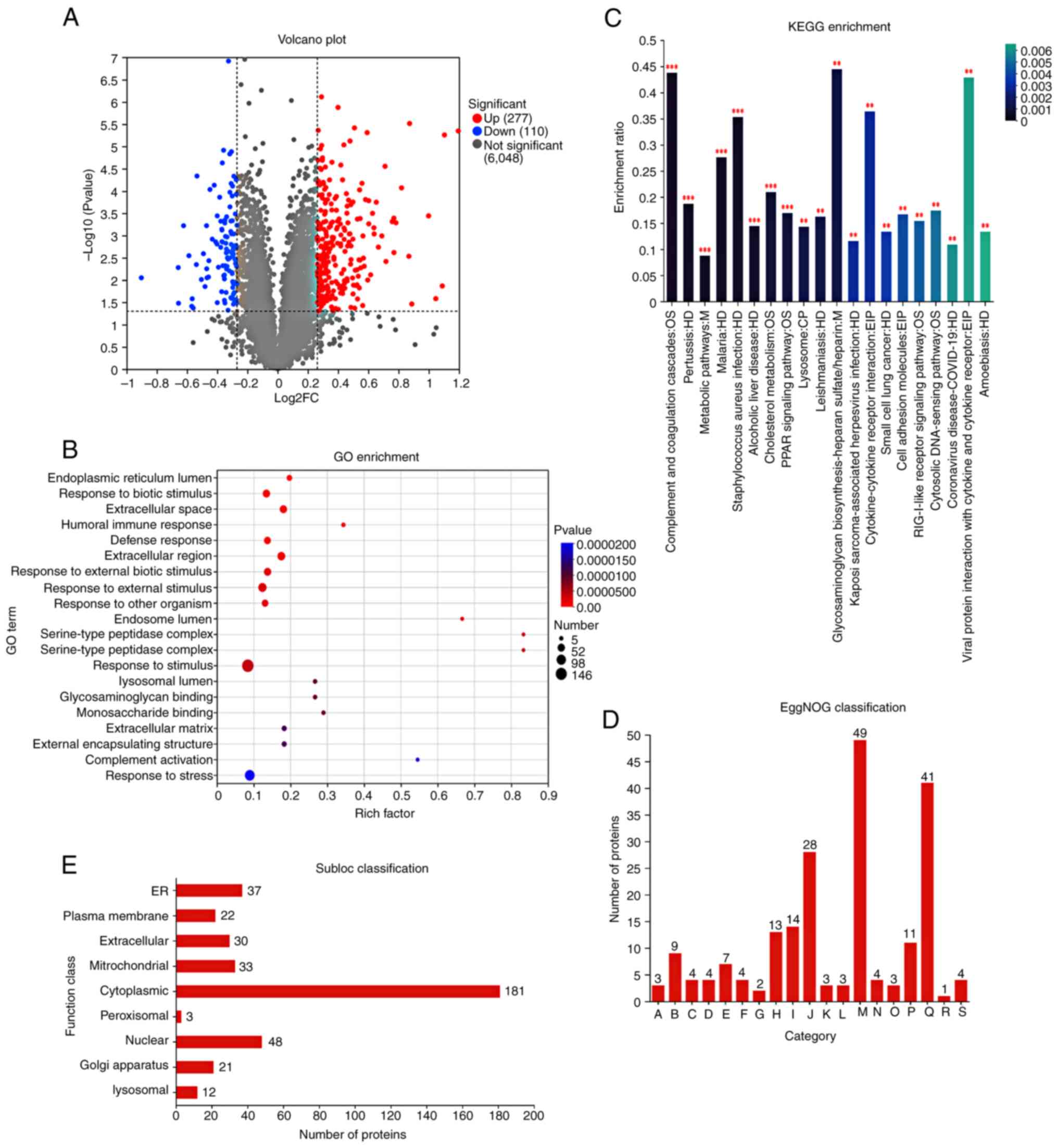 | Figure 3.Proteomics analysis of MHCC-97H cells
treated with 125I. (A) Volcano plot of differentially
expressed genes. (B) GO enrichment analysis. (C) KEGG pathway
analysis of proteomics data. (D) EggNOG classification; A, RNA
processing and modification; B, Energy production and conversion;
C, Cell cycle control, cell division, chromosome partitioning; D,
Amino acid transport and metabolism; E, Nucleotide transport and
metabolism; F, Carbohydrate transport and metabolism; G, Coenzyme
transport and metabolism; H, Lipid transport and metabolism; I,
Translation, ribosomal structure and biogenesis; J, Transcription;
K, Replication, recombination and repair; L, Cell
wall/membrane/envelope biogenesis; M, Posttranslational
modification, protein turnover, chaperones; N, Inorganic ion
transport and metabolism; O, Secondary metabolites biosynthesis,
transport and catabolism; P, Signal transduction mechanisms; Q,
Intracellular trafficking, secretion, and vesicular transport; R,
Defense mechanisms; S, Cytoskeleton. (E) Subloc classification. In
Fig. 3, **P<0.01,
***P<0.001. FC, fold change; GO, Gene Ontology; 125I,
iodine-125; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
Integrated proteomics and
transcriptomics analysis
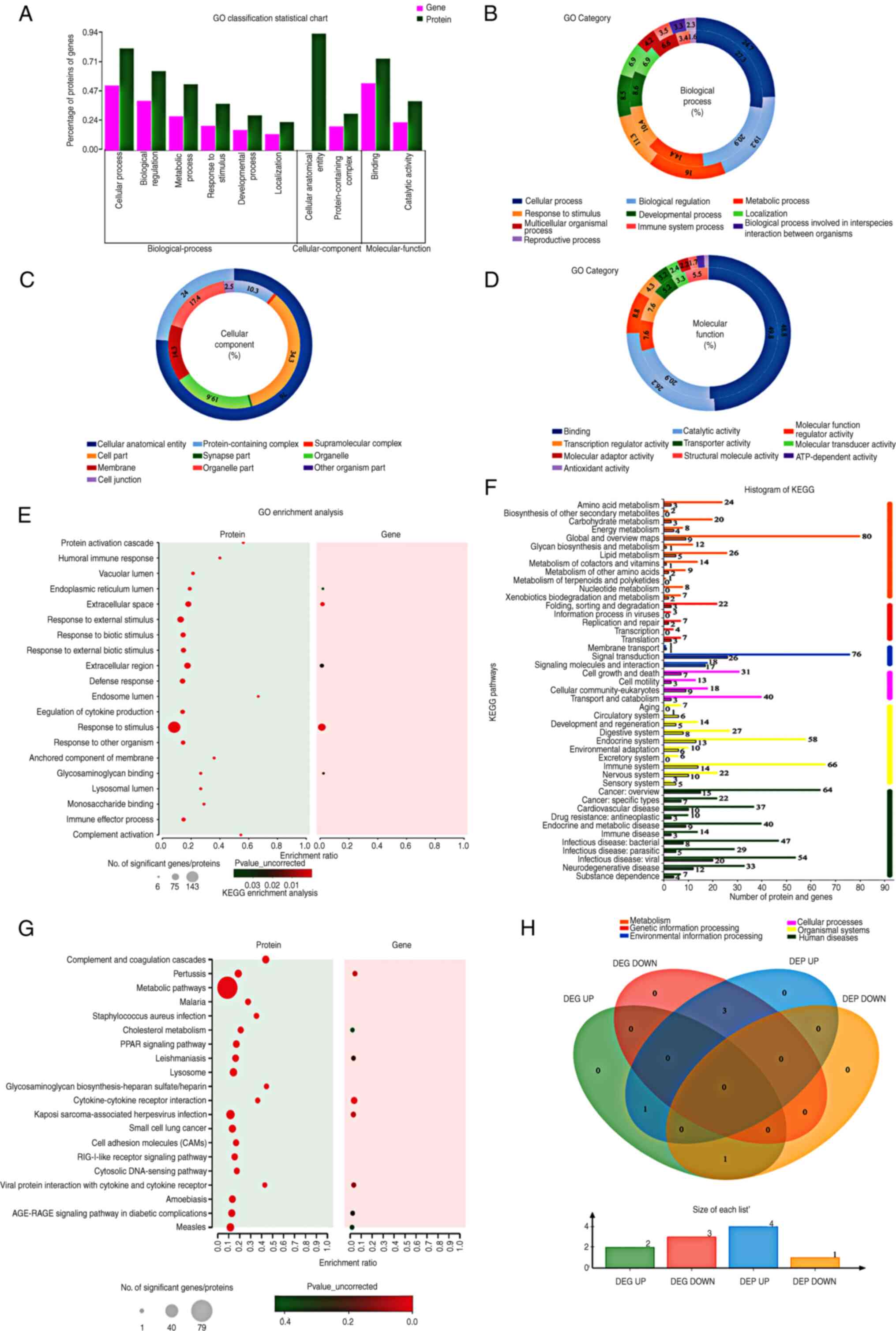
GO classification statistics revealed that the
differentially expressed genes and proteins were mainly enriched in
BP terms, followed by CC and MF terms (Fig. 4A). Regarding BP terms, genes and
proteins were predominantly distributed in cellular processes,
biological regulation and metabolic processes (Fig. 4B). Regarding CC terms, genes were
mainly distributed in cell parts and organelles, whereas proteins
were primarily located in cellular anatomical entities and in
complexes containing proteins (Fig.
4C). Regarding MF terms, both genes and proteins were mainly
distributed in binding and catalytic activities (Fig. 4D). GO enrichment analysis revealed
that genes were mainly enriched in the extracellular space,
stimulus response and glycosaminoglycan binding, whereas proteins
were mainly enriched in endosomes, protein activation cascades and
complement activation (Fig. 4E).
The KEGG histogram showed that the genes and proteins were mainly
distributed in metabolism, followed by organismal systems and human
diseases (Fig. 4F). Regarding
metabolism, genes were mainly distributed in global and overview
maps, as well as in lipid, energy and amino acid metabolism. The
proteins were primarily distributed in global and overview maps,
lipid metabolism and amino acid metabolism. Regarding organismal
systems, genes were mainly distributed in the immune, endocrine and
nervous systems, whereas proteins were primarily distributed in the
immune, endocrine and digestive systems. Regarding human diseases,
changes in genes after 125I intervention were associated
with viral/bacterial infectious diseases, cancer: overview, and
neurodegenerative disease, leading to changes in proteins related
to overview cancer, viral infectious diseases and bacterial
infectious diseases. KEGG enrichment analysis indicated that the
differentially expressed genes and proteins were mainly involved in
cytokine-receptor interactions, pertussis and Kaposi's
sarcoma-associated herpes virus infection (Fig. 4G). Transcriptomics and proteomics
data were used to describe the relationships between proteins and
genes. The Venn diagram showed a total of 5 proteins with
differential expression at both the mRNA and protein levels
(Fig. 4H).
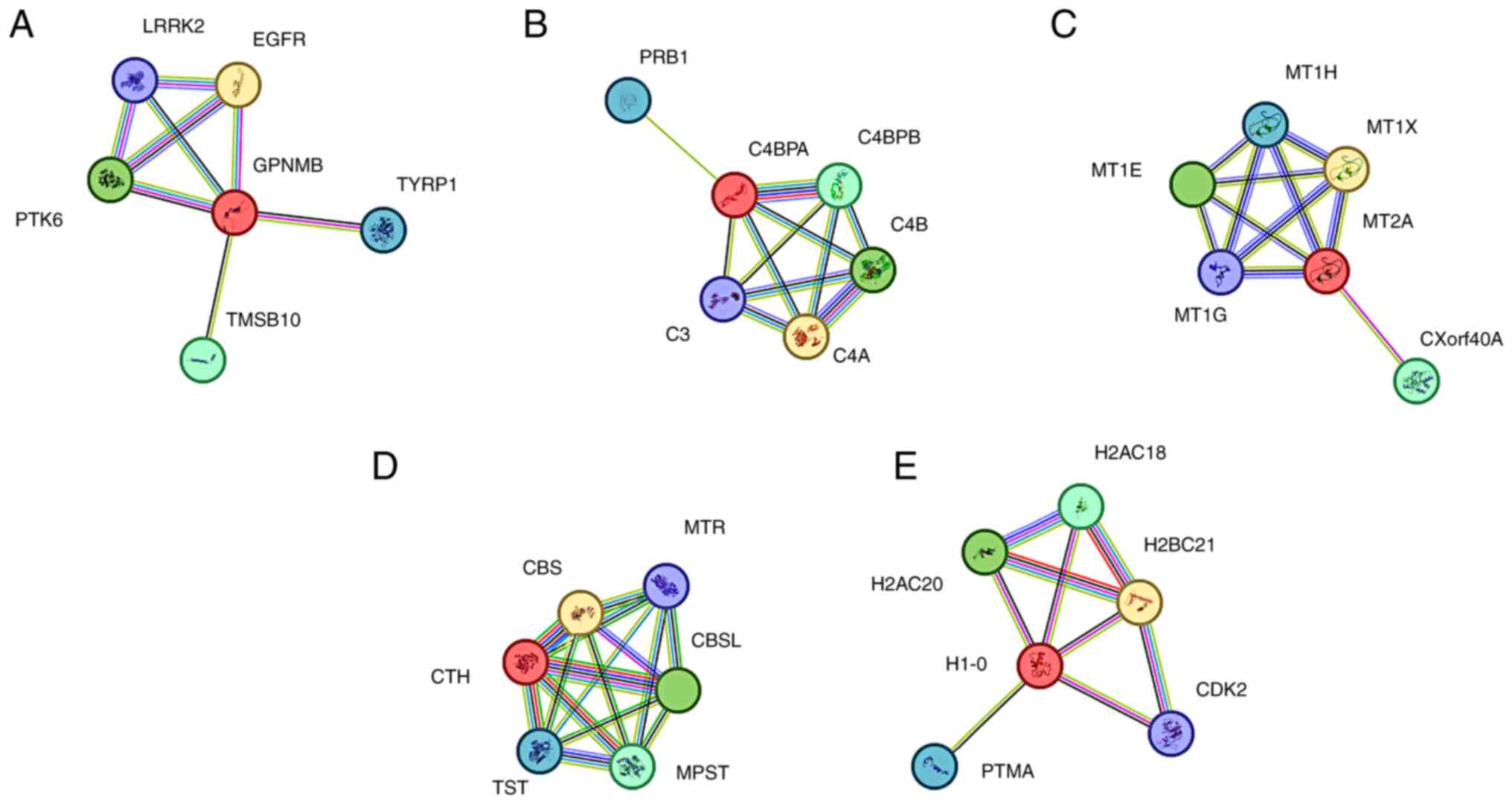
STRING analysis reveals an interaction
network of differentially expressed genes in MHCC-97H cells after
125I intervention
STRING analysis (https://string-db.org/) was used to generate a network
of the interactions between differentially expressed genes (GPNMB,
C4BPA, MT2A, CTH and H1-0) in MHCC-97H cells after 125I
intervention, highlighting their close interactions (Fig. 5A-E). GPNMB was highly expressed at
both the transcriptomic and proteomic levels in MHCC-97H cells
after 125I treatment; GPNMB may be associated with
growth delay and reduced metabolic potential. In the network
diagram, the interaction network has 6 nodes, 8 edges, an average
node degree of 2.67, an average local clustering coefficient of
0.883, 6 expected edges, and a PPI enrichment P-value of 0.223
(Fig. 5A). C4BPA, CTH and H1-0
showed low transcript and high protein expression levels after
125I intervention in MHCC-97H cells. C4BPA is involved
in the positive regulation of the complement cascade, immune
response, lectin-induced complement pathway and apoptotic cell
clearance. The interaction network of this network diagram had 6
nodes, 11 edges, an average node degree of 3.67, an average local
clustering coefficient of 0.933, an expected number of edges of 5,
and a PPI enrichment P-value of 0.0165 (Fig. 5B). CTH is involved in sulfur amino
acid metabolism, one-carbon metabolism and other related pathways.
The network diagram showed an interaction network with 6 nodes, 15
edges, an average node degree of 5, an average local clustering
coefficient of 1, an expected number of edges of 5, and a PPI
enrichment P-value of 0.000247 (Fig.
5D). H1-0 is involved in the response to stimuli and programmed
cell death pathways. The interaction network of the H1-0 network
diagram had 6 nodes, 9 edges, an average node degree of 3, an
average local clustering coefficient of 0.844, 7 expected edges,
and a PPI enrichment P-value of 0.275 (Fig. 5E). MT2A in MHCC-97H cells showed
high expression at the transcriptional level and low expression at
the protein level after 125I treatment. It is involved
in metal ion SLC transport and interferon-γ signaling. The
interaction network of this network diagram had 6 nodes, 11 edges,
an average node degree of 3.67, an average local clustering
coefficient of 0.933, an expected number of edges of 5 and a PPI
enrichment P-value of 0.0138 (Fig.
5C).
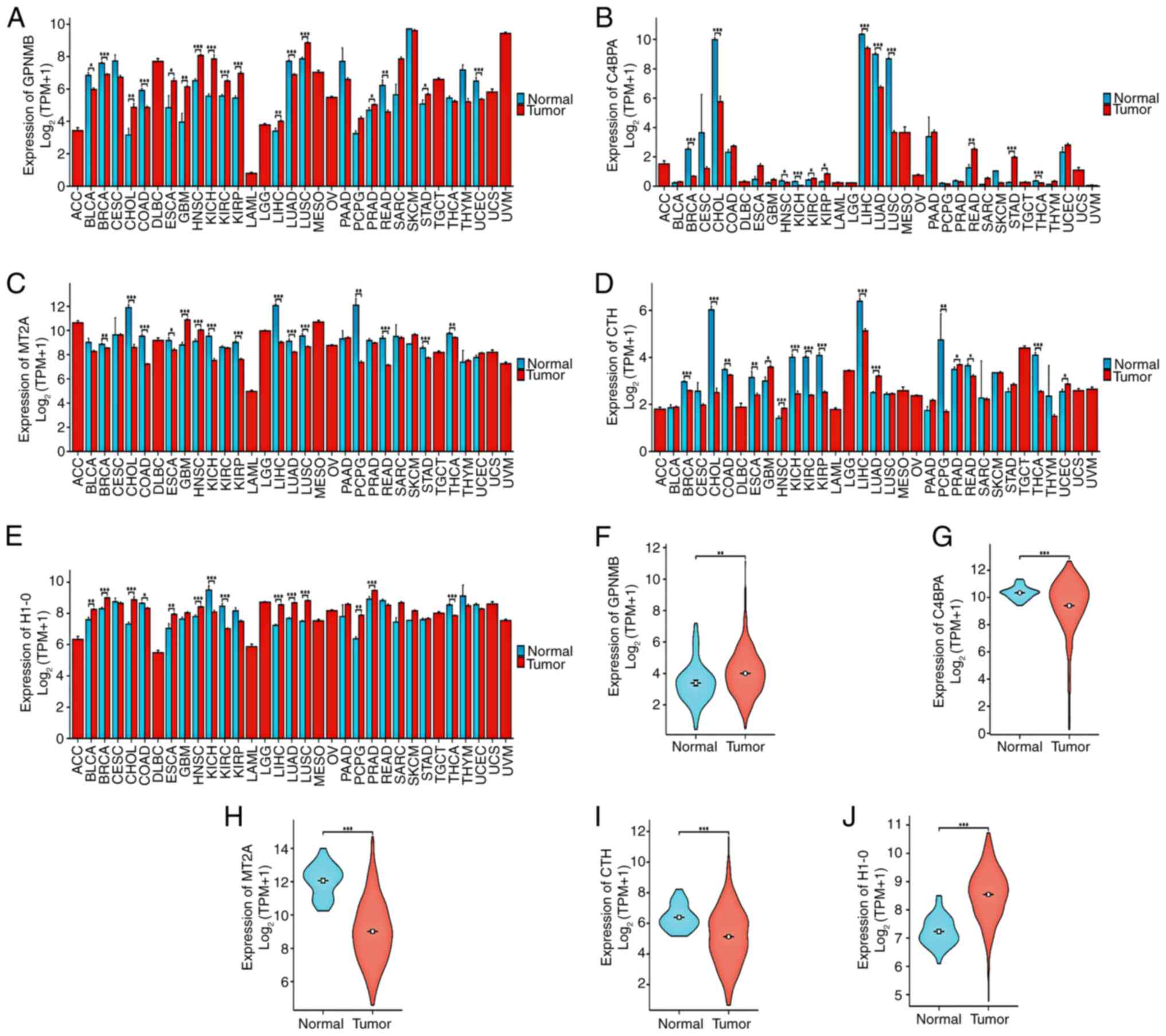
Pan-cancer analysis of differentially
expressed genes
Xiantao predicts the differential expression of
genes (GPNMB, C4BPA, MT2A, CTH and H1-0) in cancer. In the
pan-cancer analysis, the expression levels of GPNMB were
significantly upregulated in various types of cancer, particularly
in lung and kidney cancer (Fig.
6A). The expression levels of C4BPA were downregulated various
types of cancer (Fig. 6B), whereas
the expression levels of MT2A, CTH and H1-0 varied among different
types of cancer (Fig. 6C-E).
Notably, it was predicted that the expression levels of GPNMB
(Fig. 6F) and H1-0 (Fig. 6J) would be significantly higher,
whereas the expression levels of C4BPA (Fig. 6G), MT2A (Fig. 6H) and CTH (Fig. 6I) would be significantly lower in
HCC tissues compared with those in normal tissues (adjacent healthy
tissues).
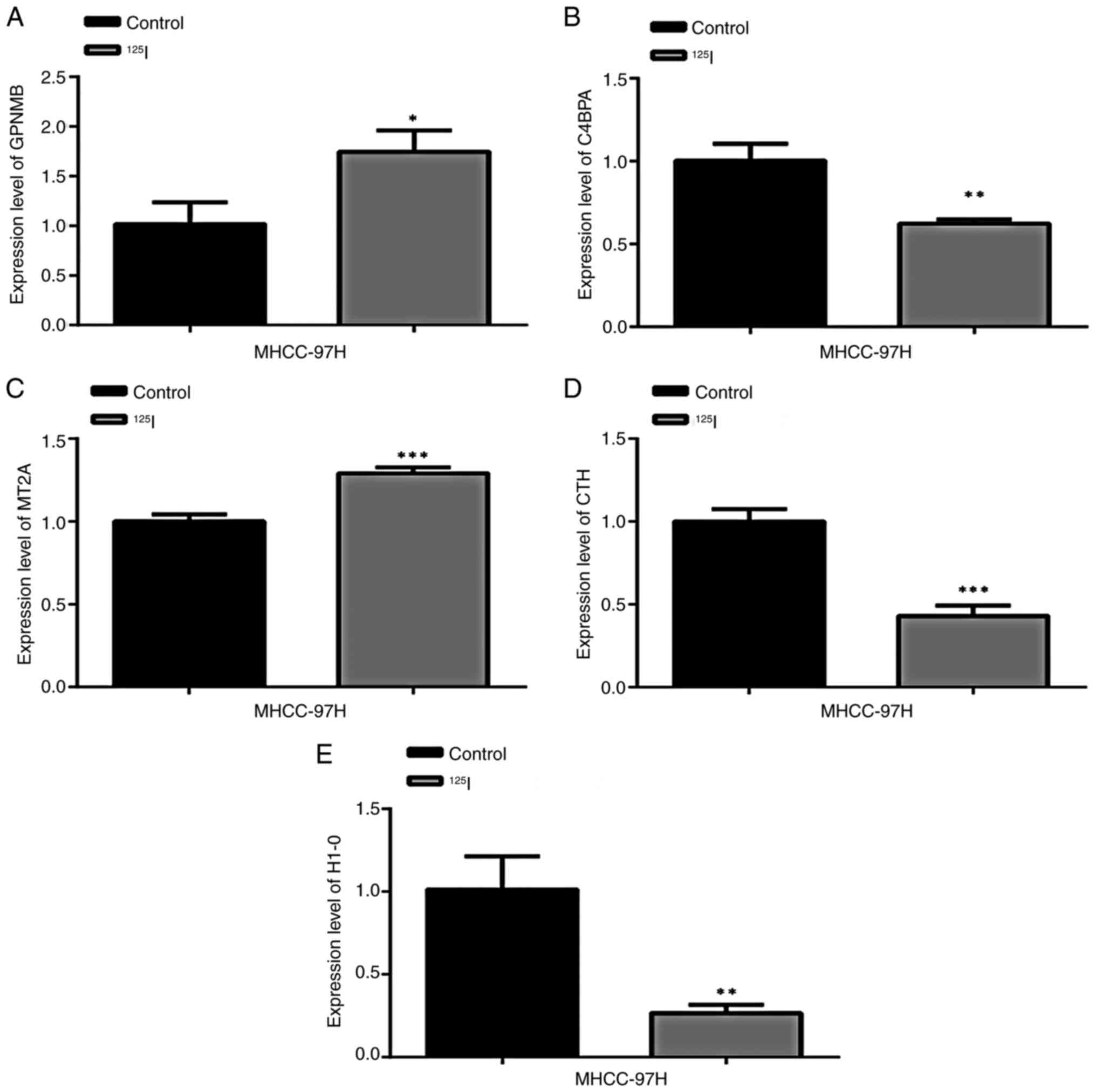
Differential gene expression
validation
RT-qPCR was used to detect the mRNA expression
levels of GPNMB, C4BPA, MT2A, CTH and H1-0 in MHCC-97 cells. The
results showed that compared with in the control group, the
expression levels of GPNMB (Fig.
7A) and MT2A (Fig. 7C) were
significantly increased in cells treated with 125I,
whereas the expression levels of C4BPA (Fig. 7B), CTH (Fig. 7D) and H1-0 (Fig. 7E) were significantly reduced.
Discussion
Liver cancer is known for its rapid cell
proliferation, invasion and metastatic capabilities. Apoptosis is a
process of programmed cell death that is closely linked to the
proliferative abilities of cancer cells and is key to controlling
tumor growth. In recent years, transcriptomics and proteomics are
emerging research fields, which have served an important role in
the study of liver cancer. Based on proteogenomic characteristics,
liver cancer can be precisely classified and targeted for
treatment, with proteins being the main units that perform cellular
functions (15). Integrating
transcriptomics and proteomics data may provide a more
comprehensive molecular landscape of liver cancer, helping to
understand how changes in gene expression translate into changes at
the protein level and affect the biological behavior of tumors,
thus offering new perspectives for the early diagnosis and
treatment of liver cancer (16,17).
The clinical application of 125I
radioactive seed implantation therapy has made significant progress
in the treatment of liver cancer. For example, CT-guided
125I close-range radiotherapy is considered a safe and
effective therapy, without serious adverse events, which has
advantages of a high local control rate and being minimally
invasive (18). The present study
indicated that 125I treatment promoted the apoptosis of
HCC cells, and inhibited their proliferation, invasion and
migration. However, the underlying mechanisms by which
125I inhibits tumor progression are not yet clear. The
present study conducted a comprehensive analysis based on
transcriptomics and proteomics data, and identified differential
expression of the mRNA and protein levels of GPNMB, C4BPA, H1-0,
CTH and MT2A in HCC cells treated with 125I. GPNMB is a
transmembrane glycoprotein that includes a long extracellular
domain (ECD) and a short intracellular domain (ICD). The ICD region
contains immunoreceptor tyrosine-based activation motifs and
leucine-rich motifs, which are associated with intracellular signal
transduction and the induction of cancer stem cell characteristics
(19,20). The ECD is involved in the
regulation of various signaling pathways, such as the
AKT-POU2F1-ECD pathway, the RB/E2F pathway and GLUT4-dependent
glycolysis, which are related to cancer cell migration, growth,
differentiation and other functions. Chen et al (21) revealed that GPNMB was highly
expressed in HCC cells, whereas inhibiting the expression of GPNMB
could reduce the progression of HCC. The present study revealed
that, after 125I intervention, GPNMB was significantly
increased at both the transcriptional and protein levels,
suggesting that GPNMB may not be a direct target of 125I
treatment, and the ECD of GPNMB remains intact after
125I treatment.
C4BPA is an effective soluble inhibitor of the
classical and lectin pathways of the complement system, composed of
complement control protein domains (22). The lack of a C4b binding site leads
to the loss of all inhibitory functions of C4BPA in the classical
complement pathway (23). Feng
et al (24) reported that
C4BPA was significantly upregulated in liver cancer tissues
compared with in adjacent healthy tissues, providing a mechanism
for cancer cells to evade immune system attacks. Inhibiting the
expression of C4BPA or blocking its interaction with the complement
system may also help to enhance the response of patients with liver
cancer to immunotherapy, thereby improving the therapeutic effect.
In the present study, after 125I intervention, the
transcriptional levels of C4BPA were significantly decreased, which
is similar to previous studies (24–26);
therefore, inhibiting the expression of C4BPA may affect the
phenotype of liver cancer cells. However, proteomics analysis
revealed that the protein expression levels of C4BPA were
significantly increased, suggesting that this discrepancy in
protein levels may be due to post-transcriptional regulation, such
as mechanisms mediated by small RNAs. Similar results were revealed
regarding the H1-0 gene. Notably, elevated expression levels of
H1-0 have been shown to be positively associated with cancer
recurrence and lower survival rates (27,28).
In paclitaxel-resistant ovarian cancer cells, H1-0 has been
reported to be upregulated, whereas knocking out H1-0 was shown to
significantly downregulate the androgen receptor, enhancing the
sensitivity of paclitaxel-resistant cell lines to paclitaxel
(27,28).
CTH, also known as CSE, is one of the key enzymes in
the production of hydrogen sulfide, which has a role in promoting
tumor formation by regulating angiogenic mechanisms in tumors. Pan
et al (29) revealed that
the expression levels of CSE were abnormally high in HepG2 and
PLC/PRF/5 liver cancer cell lines. Inhibition of CSE and its
downstream signaling pathways could activate the
mitochondrial-mediated apoptosis process and block the signal
transduction of cell proliferation, thereby inhibiting the
proliferation of liver cancer cells. In the present study, after
treatment with 125I, the mRNA expression levels of CTH
were significantly decreased, which is consistent with the results
of Pan et al (29).
However, Xiang et al (30)
recently reported that, in HCC, the expression levels of CTH were
significantly reduced, and high expression of CTH was revealed to
be associated with the active state of various immune cells. High
expression of CTH may thus be related to a better prognosis for
patients with HCC. Through proteomics analysis, the present study
revealed that, after intervention with 125I, the protein
expression levels of CTH were significantly increased, which is in
contrast to the findings of the transcriptomics analysis. The
discrepancies observed in previous studies, as well as the
inconsistency in the expression of CTH at the transcriptional and
protein levels in the present study, indicated that the specific
role and mechanism of CTH in liver cancer remain controversial.
Therefore, to fully understand the role of CTH in the development
of liver cancer, more in-depth research is needed in different
liver cancer cell lines and clinical samples. This will help to
reveal the complex mechanisms of CTH in liver cancer and to assess
its potential as a therapeutic target. Future research may need to
focus on the regulatory network of CTH and how it affects the
response of liver cancer cells to treatment and its interaction
with the immune microenvironment.
MT2A is a protein belonging to the metallothionein
family. In co-expression gene analysis of microarray data in HCC,
the transcription factor FOS and its target gene MT2A were both
revealed to be upregulated in the HCC cell line HepG2, having an
important role in the pathogenesis of HCC and potentially serving
as a therapeutic target for HCC (31,32).
According to a previous study, the levels of MT-1 and MT-2A have
been reported to be markedly reduced in primary human HCC and
diethylnitrosamine-induced mouse liver tumors, mainly due to
transcriptional repression (33).
In addition, it has been reported that in HCC the expression levels
of MT-1 and MT-2 in the nucleus and cytoplasm are closely related
to the occurrence of liver cancer and the degree of tumor
differentiation and invasiveness, and may be important biomarkers
for predicting the prognosis of patients with HCC (34). In colorectal cancer, the inhibition
of MAT2A/MAT2B expression has been shown to inhibit the migration
and invasion of cancer cells (35). In the present study, it was
revealed that the protein expression of MAT2A in liver cancer cells
was significantly decreased after 125I intervention,
which is consistent with previous research results. Therefore, it
could be hypothesized that 125I may inhibit the
proliferation, invasion and migration of liver cancer cells by
regulating the expression of the MAT2A protein. The increase in its
transcriptional mRNA levels and the decrease in its protein
expression levels after 125I intervention may be due to
the regulation of the translation process, leading to a reduction
in protein synthesis, but further experimental verification is
required.
To the best of our knowledge, the present study is
the first to link 125I intervention with changes in
specific gene expression patterns, providing new insights into the
potential roles of CTH and MT2A in HCC. Additionally, the study
explored the possible impact of these gene expression changes on
disease progression and response to treatment. However, there are
some limitations in the current study. Although the integrated
analysis revealed differential expression of five proteins at both
the mRNA and protein levels, these identified proteins lack a
protein-protein interaction network, highlighting the limitations
of our current understanding. Furthermore, the study was conducted
using a single HCC cell line, necessitating further experiments to
validate the results of the omics analysis and elucidate the
specific mechanisms of 125I action. Addressing these
issues in future research will enhance the robustness and
applicability of the study results.
In conclusion, the present study identified the
inhibitory effect of 125I on HCC, manifested as
suppression of proliferation, invasion and migration, and promotion
of cell apoptosis. The integration of transcriptomics and
proteomics data implicated MT2A and CTH in the antitumor effects of
125I. RT-qPCR validated some of the results. However,
further investigation is required to ascertain whether MT2A and CTH
act as pivotal mediators in the suppression of tumor progression by
125I. The present findings provide novel insights into
the potential mechanisms of 125I radiation particle
therapy in liver cancer, and offer new therapeutic targets for the
management of this disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the Tianjin Health Science and
Technology Project (grant no. TJWJ2021MS013) and by the Tianjin
Applied Basic Research Diversified Investment Fund Project (grant
no. 21JCYBJC01060).
Availability of data and materials
The transcriptomic data generated in the present
study may be found in the NCBI Sequence Read Archive by using the
following URL: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1135734.
The proteomic data generated in this study can be found in the
iprox with login number PXD054130 or by visiting the following
website: https://www.iprox.cn/page/PSV023.html;?url=17294821861659WCR,
password: Oqts. Other data generated in this study can be requested
from the corresponding author.
Authors' contributions
JS and ED conceived and designed the study. YY
participated in the research design, conducted experiments and
wrote the manuscript. WY assisted with the experiments, analyzed
the data and revised the manuscript. YY and JS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tsuchiya N, Sawada Y, Endo I, Saito K,
Uemura Y and Nakatsura T: Biomarkers for the early diagnosis of
hepatocellular carcinoma. World J Gastroenterol. 21:10573–10583.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang W and Wei C: Advances in the early
diagnosis of hepatocellular carcinoma. Genes Dis. 7:308–319. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang S, Cao Y, Wang R, Liu H, Wang T and
Yang S: Feasibility of 125I brachytherapy combined with
arterial infusion chemotherapy in patients with advanced pancreatic
cancer. Medicine (Baltimore). 102:e350332023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X and Wang D: Clinical analysis of
125I seed implantation combined with epidermal growth
factor receptor-tyrosine kinase inhibitors in advanced non-small
cell lung cancer. J BUON. 26:1879–1886. 2021.PubMed/NCBI
|
|
5
|
Peng S, Yang QX, Zhang T, Lu MJ, Yang G,
Liu ZY, Zhang R and Zhang FJ: Lobaplatin-TACE combined with
radioactive 125I seed implantation for treatment of
primary hepatocellular carcinoma. Asian Pac J Cancer Prev.
15:5155–5160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun H, Zhang M, Liu R, Liu Y, Hou Y and Wu
C: Endovascular implantation of 125I seed combined with
transcatheter arterial chemoembolization for unresectable
hepatocellular carcinoma. Future Oncol. 14:1165–1176. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni
A, Kamel IR, Cloyd JM and Pawlik TM: Management of hepatocellular
carcinoma: A review. JAMA Surg. 158:410–420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Juaid N, Amin A, Abdalla A, Reese K,
Alamri Z, Moulay M, Abdu S and Miled N: Anti-hepatocellular
carcinoma biomolecules: molecular targets insights. Int J Mol.
22:107742021. View Article : Google Scholar
|
|
9
|
Li D, Wang WJ, Wang YZ, Wang YB and Li YL:
Lobaplatin promotes 125I-induced apoptosis and inhibition of
proliferation in hepatocellular carcinoma by upregulating
PERK-eIF2α-ATF4-CHOP pathway. Cell Death Dis. 10:7442019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chelakkot C, Chelakkot VS, Shin Y and Song
K: Modulating glycolysis to improve cancer therapy. Int J Mol Sci.
24:26062023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu LX, Heng JH, Deng DX, Zhao H, Zheng
ZY, Liao LD, Lin W, Xu XE, Li EM and Xu LY: Sulconazole induces
panoptosis by triggering oxidative stress and inhibiting glycolysis
to increase radiosensitivity in esophageal cancer. Mol Cell
Proteomics. 22:1005512023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng J, Luo J, Zeng H, Guo L and Shao G:
125I suppressed the Warburg effect viaregulating
miR-338/PFKL axis in hepatocellular carcinoma. Biomed Pharmacother.
119:1094022019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Ma D, Wang C, Zhao S and Liu C:
Triptolide Inhibits Invasion and Tumorigenesis of Hepatocellular
Carcinoma MHCC-97H Cells Through NF-κB Signaling. Med Sci Monit.
22:1827–1836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
Relative Gene Expression Data Using Real-Time Quantitative PCR and
the 2 (−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fujita M, Chen MM, Siwak DR, Sasagawa S,
Oosawa-Tatsuguchi A, Arihiro K, Ono A, Miura R, Maejima K, Aikata
H, et al: Proteo-genomic characterization of virus-associated liver
cancers reveals potential subtypes and therapeutic targets. Nat
Commun. 13:64812022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang W, Yang C, Hu Y, Yi K, Xiao W, Xu X
and Chen Z: Comprehensive analysis of the correlation of the
pan-cancer gene HAUS5 with prognosis and immune infiltration in
liver cancer. Sci Rep. 13:24092023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou PY, Zhou C, Gan W, Tang Z, Sun BY,
Huang JL, Liu G, Liu WR, Tian MX, Jiang XF, et al: Single-cell and
spatial architecture of primary liver cancer. Commun Biol.
6:11812023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiu Z, Yu C, Qiu X, Li Q, Li J, Chen Z,
Chang S, Zhang S, Fan G and Wang S: Safety and Efficacy of
CT-Guided Iodine-125 Brachytherapy for Portal Vein Tumor Thrombus
in Hepatocellular Carcinoma. Acad Radiol. 30 (Suppl 1):S53–S60.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saade M, Araujo de Souza G, Scavone C and
Kinoshita PF: The Role of GPNMB in Inflammation. Front Immunol.
12:6747392021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie R, Okita Y, Ichikawa Y, Fikry MA,
Huynh Dam KT, Tran STP and Kato M: Role of the kringle-like domain
in glycoprotein NMB for its tumorigenic potential. Cancer Sci.
110:2237–2246. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Shan X, Wan X, Zha W and Fan R:
HOMER3 promotes liver hepatocellular carcinoma cancer progression
by -upregulating EZH2 and mediating miR-361/GPNMB axis. Pathol Res
Pract. 254:1551502024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blom AM: A cluster of positively charged
amino acids in the alpha-chain of C4b-binding protein (C4BP) is
pivotal for the regulation of the complement system and the
interaction with bacteria. Scand J Clin Lab Invest Suppl.
233:37–49. 2000.PubMed/NCBI
|
|
23
|
Blom AM: Structural and functional studies
of complement inhibitor C4b-binding protein. Biochem Soc Trans.
30:978–982. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Feng G, Li J, Zheng M, Yang Z, Liu Y,
Zhang S, Ye L, Zhang W and Zhang X: Hepatitis B virus X protein
up-regulates C4b-binding protein α through activating transcription
factor Sp1 in protection of hepatoma cells from complement attack.
Oncotarget. 7:28013–28026. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dong W, Xia Z, Chai Z, Qiu Z, Wang X, Yang
Z, Wang J, Zhang T, Zhang Q and Jin J: Proteomic analysis of small
extracellular vesicles from the plasma of patients with
hepatocellular carcinoma. World J Surg Oncol. 20:3872022.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He X, Wang Y, Zhang W, Li H, Luo R, Zhou
Y, Liao CL, Huang H, Lv X, Xie Z and He M: Screening differential
expression of serum proteins in AFP-negative HBV-related
hepatocellular carcinoma using iTRAQ-MALDI-MS/MS. Neoplasma.
61:17–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanda T, Jiang X and Yokosuka O: Androgen
receptor signaling in hepatocellular carcinoma and pancreatic
cancers. World J Gastroenterol. 20:9229–9236. 2014.PubMed/NCBI
|
|
28
|
Kohli A, Huang SL, Chang TC, Chao CC and
Sun NK: H1.0 induces paclitaxel-resistance genes expression in
ovarian cancer cells by recruiting GCN5 and androgen receptor.
Cancer Sci. 113:2616–2626. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan Y, Ye S, Yuan D, Zhang J, Bai Y and
Shao C: Hydrogen sulfide (H2S)/cystathionine γ-lyase (CSE) pathway
contributes to the proliferation of hepatoma cells. Mutat Res.
763–764. 10–18. 2014.
|
|
30
|
Xiang J, Wu X, Liu W, Wei H, Zhu Z, Liu S,
Song C, Gu Q, Wei S and Zhang Y: Bioinformatic analyzes and
validation of cystathionine gamma-lyase as a prognostic biomarker
and related to immune infiltrates in hepatocellular carcinoma.
Heliyon. 9:e161522023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu F, Li H, Chang H, Wang J and Lu J:
Identification of hepatocellular carcinoma-associated hub genes and
pathways by integrated microarray analysis. Tumori. 101:206–214.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Jiang T, Li Z, Lu L, Zhang R,
Zhang D, Wang X and Tan J: Analysis of differentially co-expressed
genes based on microarray data of hepatocellular carcinoma.
Neoplasma. 64:216–221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Datta J, Majumder S, Kutay H, Motiwala T,
Frankel W, Costa R, Cha HC, MacDougald OA, Jacob ST and Ghoshal K:
Metallothionein expression is suppressed in primary human
hepatocellular carcinomas and is mediated through inactivation of
CCAAT/enhancer binding protein alpha by phosphatidylinositol
3-kinase signaling cascade. Cancer Res. 67:2736–2746. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park Y and Yu E: Expression of
metallothionein-1 and metallothionein-2 as a prognostic marker in
hepatocellular carcinoma. J Gastroenterol Hepatol. 28:1565–1572.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tomasi ML, Cossu C, Spissu Y, Floris A,
Ryoo M, Iglesias-Ara A, Wang Q, Pandol SJ, Bhowmick NA, Seki E, et
al: S-adenosylmethionine and methylthioadenosine inhibit cancer
metastasis by targeting microRNA 34a/b-methionine
adenosyltransferase 2A/2B axis. Oncotarget. 8:78851–78869. 2017.
View Article : Google Scholar : PubMed/NCBI
|