Introduction
Heat shock proteins (HSPs) are an endogenous protein
family associated with various cellular functions in tissues, which
serve crucial roles in maintaining protein and cellular
homeostasis, and protecting cells from damage (1–3).
HSPs were initially named after the discovery of their response to
cellular heat; however, further research has revealed that HSPs can
be activated under conditions such as hypoxia, oxidative stress,
infection and exposure to heavy metals, and that they can repair
damaged proteins and protect them from damage (4–6). The
molecular sizes of HSPs range from 10 to 150 kDa, and HSPs are
roughly divided into HSP110, HSP90, HSP70, HSP60, HSP40 and small
HSPs (sHSPs) based on their molecular weights. Notably, HSPs
participate in co-translational or post-translational protein
folding (7). The molecular sizes
of sHSPs range from 12 to 43 kDa; sHSPs with a core α-crystallin
domain include HSPB1/HSP27, HSPB2, HSPB3, HSPB4, HSPB5, HSPB6,
HSPB7, HSPB8/HSP22, HSPB9 and HSPB10 (8). sHSPs are responsible for the binding
of improperly folded protein substrates. Additionally, they can
stabilize damaged proteins by exposing hydrophobic residues, and
they further transfer these proteins to ATP-dependent chaperones or
protein degradation machines, such as proteasomes or
autophagosomes, in order to prevent protein denaturation,
misfolding and abnormal aggregation (2,9–12).
HSPB8 was first described in human melanoma cells in
2000, at which time it was identified as being an H11 protein
kinase (13). Due to its molecular
weight being ~22 kDa, HSPB8 is also referred to as HSP22 (14). Initially, the investigation into
HSP22 was limited to tumors; notably, it was demonstrated to be
highly expressed in specific tumors, such as gastric cancer
(15), breast cancer (16), ovarian cancer (17), hepatocellular carcinoma (18) and glioblastoma (19). Moreover, HSP22 has been shown to be
highly expressed in breast cancer and ovarian cancer, and to
promote the occurrence and development of tumors by regulating the
proliferation and migration of tumor cells (16,17).
Conversely, HSP22 is expressed at low levels in other tumor tissues
or cell lines, such as melanoma and prostate cancer (20,21).
By treating with demethylating agents, the apoptosis of tumor cells
with a low expression of HSP22 can be induced (20–22).
Furthermore, an exploration into sHSPs revealed that HSP22 is
mainly expressed in skeletal muscle and the myocardium (9,23).
In skeletal muscle, HSP22 may be involved in muscle development and
repair processes (24,25). In addition, mutations or the
abnormal expression of HSP22 may be closely related to the
occurrence and development of certain muscle diseases, such as
hereditary peripheral motor neuropathy (26). However, compared with in cardiac
tissue, the stress protective effect of HSP22 in skeletal muscle
may not be as significant; notably, there is evidence suggesting
that myocardial ischemia can induce the expression of HSP22 in
cardiac tissue in animal models and patients (27–29).
In addition, the upregulation of HSP22 may protect myocardial cells
from hydrogen peroxide (H2O2)-induced
oxidative stress and cell death in vitro and reduce
myocardial ischemic damage in vivo (30–33).
Notably, the downregulation of HSP22 has been reported to increase
stress-induced myocardial cell death, thus accelerating the
transition to cardiac failure during cardiac overload (34). A lack of HSP22 can also disrupt
cardiac energy metabolism homeostasis and increase oxidative
damage, thus leading to cardiac dilation and dysfunction (35). These findings indicate the
indispensable role of HSP22 in the heart; therefore, it may be
considered a novel therapeutic target for cardiovascular
diseases.
The present review aimed to extensively explore and
analyze the role and function of HSP22 in regulating the potential
mechanisms of cardiovascular disease. By systematically reviewing
existing research results in various aspects, the key role of HSP22
in cardiovascular disease at the molecular, cellular and tissue
levels is clarified, and its mechanism of impact on the occurrence
and development of cardiovascular disease is demonstrated. In
addition, the present study provides new perspectives and potential
targets for the treatment and prevention of cardiovascular diseases
by comprehensively evaluating the biological characteristics of
HSP22, and the complexity of its interaction with the
cardiovascular system.
Protective effect of HSP22 on myocardial
cells
HSP22 is a transmembrane protein with a stable
tertiary structure, which contains long α-helices and short β-curls
that result in a high degree of stability (9,14).
HSP22 is widely expressed in almost all tissues (14,36–38).
Notably, HSPs serve an important ‘housekeeping’ role; therefore,
any damage to the HSP network can be dangerous to cells. In the
heart, HSP22 has been shown to be inducible under various
endogenous or exogenous stimuli, and to be regulated by the
upregulation of heat shock factor (HSF) transcription. Studies have
shown that the activation of HSF1 and HSF2 is involved in cardiac
development and regulates the expression of HSPs in cardiovascular
diseases (39,40). In general, the transcription and
expression of HSPs are unlikely to be impacted by HSF; however,
under stress conditions, unfolded proteins may disrupt the
interaction between the two proteins, thus inducing the activation
and translocation of HSF and leading to the transcription of HSP22
(41,42). Notably, previous in vivo and
in vitro studies have demonstrated a marked increase in
HSP22 in the myocardium after acute and chronic ischemia, whereas
it is not induced in the normal myocardium (27,28).
In addition, the short-term overexpression of HSP22 adenovirus in
pig hearts can reduce infarct size after ischemia-reperfusion (I/R)
and upregulate myocardial inducible nitric oxide (NO) synthase
(iNOS) (30). These data indicate
that HSP22 is a stress-related protein that can be stimulated in
the hypoxic adaptive myocardium. The inducement of this protein may
have a cellular protective effect, thus preventing irreversible
damage to the ischemic myocardium. Herein, the present study
summarized the basic molecular mechanisms by which HSP22
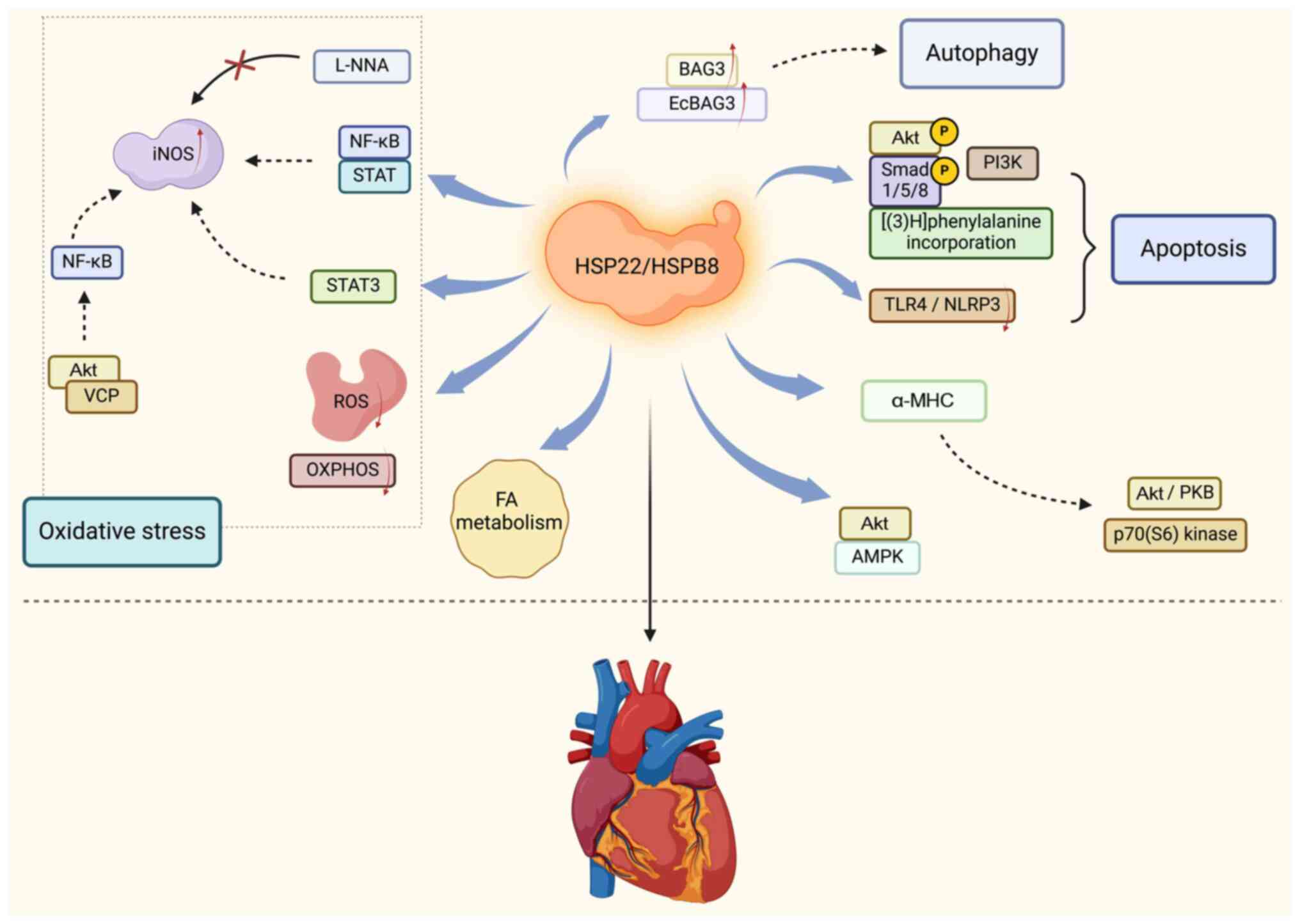
participates in regulating myocardial cell protection (Fig. 1).
HSP22 protects the heart by regulating
oxidative stress
iNOS is an enzyme that catalyzes the production of
NO from L-arginine and serves an important role in the process of
cardiac oxidative stress. Multiple studies have shown that the
cardioprotective effect of HSP22 depends on the induction of iNOS.
Depre et al (33) reported
that the overexpression of HSP22 can induce the expression of iNOS
in transgenic (TG) mice, thus serving as an effective method of
ischemic preconditioning to protect the heart. In addition, iNOS
inhibitors were shown to eliminate the cellular protective effect
of HSP22 (33). Chen et al
(30) also reported that HSP22
increases the expression of iNOS through the transcription factors
NF-κB and STAT in isolated myocardial cells. Further research has
revealed that the NO synthesis inhibitor L-NNA can inhibit the
ability of HSP22 to reduce the infarct size of pigs (30). Qiu et al (34) reported that the absence of HSP22
impairs the nuclear and mitochondrial functions of STAT3 and
reduces the expression of iNOS in cardiomyocytes under cardiac
stress, thus indirectly indicating a significant correlation
between HSP22 and iNOS expression. In addition, Lizano et al
(43) reported that the expression
of valosin-containing protein (VCP) in cardiomyocytes is highly
associated with iNOS expression; as a substrate of Akt, VCP can
mediate an increase in iNOS expression downstream of HSP22 via an
NF-κB-dependent mechanism. These findings indicate that iNOS is an
important mediator of the ability of HSP22 to protect the
heart.
Another key component of cardiac oxidative stress
involves reactive oxygen species (ROS), which are defined as
reactive molecules or free radicals containing oxygen (44). These molecules or free radicals are
usually produced during cellular metabolism and have high chemical
activity. Common ROS include superoxide anions,
H2O2, hydroxyl radicals and singlet oxygen
(45–47). Low concentrations of ROS are
involved in cellular signaling and regulation, such as cell
proliferation, differentiation and immune responses (45). However, when the generation of ROS
exceeds the antioxidant capacity of cells, it can cause oxidative
stress, thus leading to oxidative damage to intracellular
biomolecules such as DNA, proteins and lipids. Therefore, the
maintenance of a balance of ROS is crucial for health. Multiple
studies have shown that the upregulation of HSP22 in myocardial
cells can reduce H2O2-mediated cell apoptosis
by 60% (30–33). In addition, Qiu et al
(34) investigated isolated
mitochondria from the hearts of TG mice and reported that the
overexpression of HSP22 weakened hypoxia-induced oxidative
phosphorylation and ROS production. However, although HSP22 can
protect mitochondrial function under stress conditions, long-term
chronic overexpression may interfere with the normal function of
mitochondria. Morin et al (48) reported that long-term chronic
overexpression of HSP22 may affect the process of mitochondrial
oxidative phosphorylation, thus leading to reduced ATP synthesis
and increased ROS generation. The excessive production of ROS can
cause damage to mitochondria, thus further exacerbating
mitochondrial dysfunction and affecting the normal physiological
function of the heart (49). When
the heart is subjected to long-term pressure or injury stimulation,
myocardial cells undergo hypertrophy to compensate for changes in
heart function. HSP22 may be involved in this process; moreover, if
it is upregulated for a long period of time, it may continue the
process of myocardial hypertrophy and lose normal compensatory
balance (48). Cardiac hypertrophy
and fibrosis can cause a decrease in the elasticity and compliance
of the heart, thus affecting its diastolic function and ultimately
leading to heart failure. In addition, long-term chronic
upregulation of HSP22 may accelerate the aging process of the heart
(50). This effect may be related
to the interference of HSP22 in mitochondrial function, as
mitochondria are key organelles for cellular energy metabolism and
signal transduction, and their dysfunction can lead to cellular
aging and functional decline. The electrical activity of myocardial
cells depends on the normal opening and closing of various ion
channels, such as sodium, potassium and calcium channels (51). HSP22 may interact with ion channel
proteins; additionally, when it is upregulated for a long period of
time, it may alter the expression level, distribution or functional
characteristics of ion channels, thereby affecting the metabolic
processes of the heart (51).
These studies emphasize the importance of HSP22 in regulating
oxidative stress in cardiomyocytes.
HSP22 promotes cardiac autophagy and
antiapoptotic effects
Autophagy is an important process in which cells
clear damaged or excess organelles, proteins and other cellular
components, which is crucial for maintaining cellular homeostasis
and function (52–54). In the heart, autophagy helps to
clear damaged mitochondria and proteins, thus preventing the
accumulation of intracellular waste. HSP22 can protect myocardial
cells from stressful stimuli (such as oxidative stress, I/R injury
and toxins) by promoting autophagy, thereby increasing the
tolerance and function of the heart (55–60).
Bcl-2-associated homolog 3 (BAG3) is a cochaperone protein that is
highly expressed in the heart, and extensive research has shown
that HSP22 is functionally dependent on BAG3 (61–63).
Arndt et al (61) and Carra
et al (63) reported that
HSP22 and its copartner BAG3 serve crucial roles in cardiac
autophagy. HSP22 is responsible for identifying misfolded proteins,
and BAG3 (at least partially through its proline-rich domain) may
recruit and activate macrophage autophagy mechanisms that tightly
bind to chaperone molecular substrates. Depre et al
(33) also demonstrated that the
synergistic effect of BAG3 and HSP22 induces autophagic
degradation. Wu et al (35)
reported that the absence of HSP22 can impair BAG3 expression and
associated cardiac autophagy, disrupt cardiac energy metabolism
homeostasis and increase oxidative damage. Liang et al
(64) reported that the
upregulation of HSP22 can increase the expression of EcBAG3 (a
homolog of BAG3), whereas the knockdown of EcBAG3 can affect
autophagy. These findings indicate that HSP22 has a crucial role in
cardiac autophagy.
By regulating autophagy, HSP22 can reduce the
activation of apoptosis-related proteins (such as Bax), thereby
reducing myocardial cell apoptosis, which is particularly important
under pathological cardiac conditions (64,65).
Akt is a downstream effector of PI3K. Sui et al (31) reported that the upregulation of
HSP22 increases PI3K activity, Akt phosphorylation, Smad 1/5/8
phosphorylation and [(3)H]
phenylalanine incorporation, as well as causing a 70% reduction in
cell apoptosis mediated by H2O2 in isolated
muscle cells. Lan et al (66) reported that the upregulation of
HSP22 in the heart can inhibit doxorubicin-induced cardiac injury,
alleviate inflammation and reduce cardiac apoptosis by blocking
TLR4/NLRP3 activation. Moreover, Yu et al (50) demonstrated that HSP22 can improve
lipopolysaccharide-induced myocardial injury by inhibiting
inflammation, oxidative stress and cell apoptosis. By contrast, a
previous study suggested that HSP22 has dose-dependent and dual
hypertrophic and proapoptotic functions in myocardial cells
(29). In the cardiac myocytes of
rats, HSP22 can induce hypertrophy at low doses via
kinase-independent Akt activation, whereas at high doses, it can
induce cell apoptosis via a protein kinase-dependent mechanism,
particularly by inhibiting casein kinase 2 through interaction with
this kinase and subsequent physical interactions (29).
HSP22 is responsible for subcellular
redistribution
HSP22 is not only a companion protein but is also
responsible for the subcellular redistribution of other proteins.
Depre et al (67) reported
that the upregulation of HSP22 increases the protein/DNA ratio in
isolated neonatal rat cardiomyocytes by 37%. In this previous
study, a heart-specific transgene of HSP22 driven by the α-MHC
promoter was generated, thus resulting in an average 7-fold
increase in HSP22 expression, accompanied by dose-dependent
activation of the kinases Akt/PKB and p70 (S6) (67). Qiu et al (34) reported that HSP22 promotes STAT3
translocation to mitochondria, regulates mitochondrial function in
isolated neonatal rat cardiomyocytes, and increases oxidative
phosphorylation. Rashed et al (68) also demonstrated that the
translocation of HSP22 and iNOS to the mitochondria is necessary
for HSP22-mediated oxidative phosphorylation. HSP22 mainly
aggregates in the perinuclear compartment and mitochondrial inner
membrane, which provides the foundation for its interaction with
other proteins to enter into the nucleus and mitochondria.
HSP22 enhances proteasome
activity
Multiple studies have shown that HSP22 is closely
related to the expression of proteasomes. A pioneering study by
Depre et al (33) revealed
that HSP22 can participate in the metabolic switch mechanism of
ischemic hearts by promoting the activity of 5′ AMP-activated
protein kinase (AMPK). HSP22 directly binds to Akt and AMPK, thus
promoting their nuclear translocation and binding to multiple
protein complexes, which stimulates the survival mechanisms of cell
solutes and nuclei, and achieves a protective effect on the heart
(33). In addition, HSP22-mediated
cardiac hypertrophy promotes increased expression and activity of
proteasomes, as well as their subcellular redistribution (69). The inhibition of proteasomes can
prevent the increase in the protein synthesis rate that is observed
after the upregulation of HSP22 or the addition of hypertrophic
stimuli, thereby reversing cardiac hypertrophy (69). Wu et al (35) reported that HSP22 deficiency
disrupts key regulatory enzymes for fatty acid (FA) transport and
FA oxidation, thus disrupting FA metabolism, and leading to cardiac
dilation and aging. These findings indicate a close relationship
between HSP22 and the energy metabolism of proteasomes. Notably, it
is still unclear as to how HSP22 regulates and enhances the
activity of proteasomes in ischemic hearts.
Regulating HSP22 to improve cardiovascular
disease
Based on the critical role of HSP22 in the heart, a
number of studies have reported on the effects of drugs or
cytokines that regulate HSP22 in cardiovascular disease.
Geranylgeranylacetone (GGA) can induce
HSP22 to protect the heart
GGA, which is also known as teprenone, has been
widely used as an oral anti-ulcer drug in Japan and China since
1984 (70). Notably, it has been
reported that GGA can induce the upregulation of HSPs under stress
conditions; however, its induction effect is weaker in the absence
of stimulation (70,71). Multiple studies have shown that GGA
serves a protective role in inflammation, ischemia, oxidative
stress and the toxin response by increasing the expression of HSPs
(72,73). In recent years, research on GGA and
cardiovascular disease has focused on atrial fibrillation (AF)
(Table I). Brundel et al
(74) reported that the
administration of heat shock and GGA before tachy-pacing in
patients with paroxysmal AF can almost completely prevent
tachy-pacing-induced myolysis. Notably, the upregulation of HSP27
may also provide complete protection against pacing-induced
myolysis, which helps to limit its progression to persistent AF
(74). Furthermore, additional
studies have shown that HSP induction can prevent remodeling caused
by tachycardia. In vitro experiments have shown that the
protective effect of an atrial cell line (HL-1) on cardiomyocytes
requires HSP27 induction and phosphorylation (75). In clinically relevant animal
models, the oral HSP inducer GGA has a protective effect on AF
(75). Sakabe et al
(76) also reported that GGA can
prevent atrial conduction abnormalities caused by ischemic
arrhythmias and inhibit ischemic AF. Zhang et al (77) demonstrated that pretreatment with
heat shock, or the HSP inducers GGA and BGP-15, can lead to
endogenous HSP upregulation and prevent tachycardia-induced
remodeling. Chang et al (78) demonstrated that GGA can reduce
triggering activity, action potential duration dispersion and AF
induction ability in the heart during heart failure by inducing
HSP, and regulating ion channels and calcium homeostasis.
Furthermore, van Marion et al (79) identified new GGA derivatives
(especially GGA * −59), which can promote HSP expression, thereby
preventing and restoring tachy-pacing-induced remodeling. Further
research has demonstrated that GGA * −59 and recombinant HSPB1 can
accelerate tachy-pacing-induced structural remodeling and the
recovery of contractile dysfunction in HL-1 cardiomyocytes
(80). GGA * −59 can also increase
HSPB1 levels after tachy-pacing, inhibit HDAC6 activity, and
restore contractile protein and microtubule levels. In addition, a
previous study reported that 3 days of GGA treatment in the right
and left atrial appendages of patients who underwent coronary
artery bypass grafting was associated with increased levels of
HSPB1 and HSPA1 expression, as well as increased levels of HSPB1 in
muscle fibers (81). In addition,
a recent study revealed that GGA can reduce myocardial cell
stiffness and alleviate diastolic dysfunction in a rat model of
metabolic syndrome by increasing myofilament binding to HSPB1 and
HSPB5 (82). Although current
research on the role of GGA in AF is detailed, reports on the
regulation of HSP22 by GGA is currently limited.
 | Table I.Function and potential mechanism of
GGA-induced HSP. |
Table I.
Function and potential mechanism of
GGA-induced HSP.
| First author,
year | Disease | Functions and
potential mechanisms of GGA-induced HSP | (Refs.) |
|---|
| Brundel, 2006; | Paroxysmal | Upregulation of
HSP27prevents myolysis and muscle remodeling caused by | (74,75) |
| Brundel, 2006 | AF | tachycardia |
|
| Sakabe, 2008 | AF | Prevents atrial
conduction abnormalities and suppresses ischemic atrial
fibrillation | (76) |
| Zhang, 2011 | AF | Promotes endogenous
HSP upregulation and prevents tachycardia remodeling | (77) |
| Chang, 2013 | Arrhythmia | Induces HSP and
regulates ion channels and calcium homeostasis, reducing triggering
activity, action potential duration dispersion, and AF induction
ability in heart failure | (78) |
| van Marion,
2019 | AF | Promotes HSP
expression to prevent and restore remodeling induced by
tachypacing | (79) |
| Hu, 2019 | AF | Increased HSPB1
levels inhibit HDAC6 activity and restore contractile protein and
microtubule levels | (80) |
| van Marion,
2020 | Undergoing | GGA treatment is
related to higher HSP27 and HSP70 expression levels in | (81) |
|
| CABG | RAA and LAA of
patients undergoing CABG surgery, and higher HSP27 levels in the
myofilaments |
|
| Waddingham,
2023 | Cardiac metabolic
syndrome | Reduces myocardial
cell stiffness and alleviates diastolic dysfunction by increasing
the binding of HSPB1 and HSPB5 to myofilaments | (82) |
In 2009, Sanbe et al (83) first proposed that the oral
administration of GGA can increase the expression levels of HSP22
and HSPB1 in the hearts of HSPB5 R120G TG (R120G TG) mice. Compared
with in untreated R120G TG mice, R120G TG mice treated with GGA
exhibited reduced heart size and interstitial fibrosis, as well as
improved cardiac function and survival rates. In addition,
heart-specific TG mice expressing HSP22 were constructed; the
overexpression of HSP22 was shown to lead to a decrease in the
formation of amyloid oligomers and aggregates, thereby improving
cardiac function and the survival rate (83). Marunouchi et al (84) reported that treatment with GGA at
2–8 weeks after myocardial infarction in rats can alleviate the
decrease in the mitochondrial HSPB1 and HSP22 content, and that
oral administration of GGA can maintain the mitochondrial energy
production capacity and cardiac pump function during heart failure.
These findings indicate that GGA treatment may induce the
expression of sHSPs in the infarcted heart, which helps to maintain
mitochondrial function and improve cardiac contractile function.
Recently, a study published by our team revealed that GGA can
induce the expression of HSP22, protect the vascular endothelium of
mice from oxidized low-density lipoprotein damage, and block the
development of atherosclerosis (85). Although it is still unclear as to
how GGA regulates HSP22, this evidence demonstrates the significant
potential of GGA and HSP22 in cardiovascular disease (Table II).
| Table II.Medications or cytokines that control
HSP22 to treat heart conditions. |
Table II.
Medications or cytokines that control
HSP22 to treat heart conditions.
| First author,
year | Drugs or
cytokines | HSP22-associated
functions and potential mechanisms | (Refs.) |
|---|
| Sanbe, 2009 | GGA | Increasing the
expression levels of HSP22 and HSPB1 in the heart leads to a
decrease in the formation of amyloid oligomers and aggregates,
reducing heart size and interstitial fibrosis, and improving heart
function and survival rate | (83) |
| Marunouchi,
2014 | GGA | Reduces the
decrease in mitochondrial HSPB1 and HSP22 content, maintains
mitochondrial energy production capacity and cardiac pump
function | (84) |
| Gong, 2019 | GGA | Induces HSP22
expression and protects mouse vascular endothelium from ox-LDL
damage | (85) |
| Lizano, 2013 | VCP | Mediates the
increase of iNOS expression downstream of HSP22 through an NF-κB
dependent mechanism | (43) |
| Jiang, 2017 |
microRNA-126a-5p | Promotes in
vivo I/R injury by inhibiting the expression of HSP22 | (86) |
| Ren, 2020 | ZBTB20 | The expression of
HSP22 is significantly reduced in ZBTB20-null myocardium | (87) |
| Martin, 2022 | JG-98 | Reduces autophagic
flux, and alters the expression of BAG3 and HSP22 | (88) |
| Cheng, 2023 | DUSP12 | Reduces cell
apoptosis and oxidative stress in myocardial I/R injury through
HSP22-induced mitochondrial autophagy | (89) |
Other cytokines that regulate HSP22 to
improve cardiovascular disease
In addition to GGA, numerous studies have reported
the regulatory effects of other molecules on HSP22. Lizano et
al (43) reported that the Akt
substrate VCP may mediate an increase in iNOS expression downstream
of HSP22 via an NF-κB-dependent mechanism. Via bioinformatics
analysis combined with experimental validation, Jiang et al
(86) demonstrated that
microRNA-126a-5p is upregulated in myocardial I/R model mice and
promotes in vivo I/R injury by inhibiting the expression of
HSP22. Ren et al (87)
reported that the lack of the zinc finger protein ZBTB20 reduces
the cardiac ATP content, and impairs the enzyme activity of
mitochondrial complex I and the L-type calcium current density in
myocardial cells. Moreover, the expression of HSP22 was shown to be
significantly reduced in the ZBTB20-null myocardium. Martin et
al (88) reported that
treatment with JG-98 (a small-molecule therapeutic agent applied to
tumors) can reduce autophagic flux, and alter the expression of
BAG3 and several binding partners involved in BAG3-dependent
autophagy, including SYNPO2 and HSP22. In addition, a recent study
demonstrated that the upregulation of dual specificity phosphatase
12 can reduce cell apoptosis and oxidative stress in myocardial I/R
injury via HSP22-induced mitochondrial autophagy (89). Various studies have shown that
HSP22 is involved in regulating the physiological and pathological
processes of the heart, although its molecular mechanism remains
largely unknown (Table II).
Perspectives
The present study provides an overview of new
research and progress on the role of HSP22 in cardiovascular
diseases; however, several issues require further investigation.
First, as described in the present review, long-term chronic
upregulation of HSP22 in the heart may increase oxidative
phosphorylation and mitochondrial ROS production, which ultimately
leads to aging and a shortened lifespan (48). More research is still needed to
verify the potential risks of the long-term chronic upregulation of
HSP22, which will provide further insights into the efficacy and
safety of HSP22 in the heart. Second, an appropriate increase in
the expression of HSP22 within a certain range can increase the
resistance of animals to high glucose, high fat and other stress
factors, as well as protect endothelial cells from damage and
reduce the likelihood of atherosclerosis (85). Although, to the best of our
knowledge, there are currently no direct studies on the effects of
the upregulation of HSP22 in animals, in accordance with the
characteristics of the HSP family, its upregulation may lead to an
imbalance of protein homeostasis in cells, thus resulting in a
series of adverse consequences (90,91).
Therefore, in practical applications, it is necessary to carefully
control the expression levels of HSP22. Future research can explore
the specific mechanisms of the effects of different doses of HSP22
on animals, and determine how to prevent and treat related diseases
by regulating the expression of HSP22.
Third, a previous study reported that HSP22 is also
upregulated during the aging process in fruit flies (92); however, the age-related expression
of HSP22 in the heart is still debatable. Fourth, the role of
GGA-induced HSP22 in cardiovascular disease has not been well
explored and whether GGA activates HSP22 differently in various
cardiovascular disorders is still up for debate. As a HSP inducer,
GGA can trigger the expression of numerous HSPs, including HSP22,
and has broad-spectrum pharmacological effects; however, the
pathophysiology and etiology of various cardiovascular diseases
differ, which could result in variations in how GGA induces HSP22
in various conditions. To confirm the precise discrepancies, more
investigation is required. Fifth, although the cardiovascular
benefits of HSP22 have been demonstrated in animal experiments, no
relevant clinical studies are available regarding these effects.
Through large-scale clinical sample analysis, it may be possible to
identify specific biomarkers related to HSP22 function that can be
easily detected in human blood, urine or tissues. These biomarkers
could be used for early diagnosis, disease monitoring and treatment
efficacy evaluation of diseases, thus helping to screen the most
suitable patient population for HSP22-related treatment. Based on
the mechanism of action and structural characteristics of HSP22,
drugs that can specifically activate HSP22 gene transcription or
increase its protein activity can be developed in the future;
alternatively, inhibitors or agonists targeting key molecules in
the HSP22 signaling pathway can be developed. In addition to common
intravenous and oral administration methods, specific
administration methods, such as local myocardial injections and
transdermal administration, could also be explored to increase the
concentration of drugs in the heart, enhance treatment efficacy,
and reduce systemic side effects. Furthermore, a critical analysis
of the potential clinical application of HSP22 in cardiovascular
disease is required. Although HSP22 has multiple biological
functions and protective effects, its specific application value
and safety still require further verification. In future research,
a deeper understanding of the regulatory mechanisms, pathways of
action, and interactions between HSP22 and the cardiovascular
system is required to provide more accurate and effective
strategies for the treatment and prevention of cardiovascular
diseases.
Finally, a number of patients with cardiovascular
disease take medications that may cause gastrointestinal side
effects; thus, GGA (as a gastric mucosal protector) may have
potential in regulating HSP22 to improve cardiovascular disease. In
the future, the specific mechanism by which GGA affects
cardiovascular disease should be further clarified, and its role
and effects in different types of cardiovascular diseases should be
explored. In addition, no specialist research has yet been done on
how GGA interacts with other widely used medications to treat heart
disease. The safety and efficacy of GGA in combination with other
cardiovascular drugs warrant further investigation. In the future,
by measuring the concentration and time curve of GGA and its
metabolites in the body, the metabolic process and characteristics
of GGA in the body can be understood, thus providing a basis for
formulating reasonable medication plans. By combining both in
vitro and in vivo experiments, the interactions between
GGA and other heart disease treatment drugs, including metabolic
and pharmacological interactions, can be investigated. In addition,
research in genetics and pharmacology can be used to understand the
differences in response to GGA among different patients and the
reasons for these differences, thus providing a scientific basis
for personalized medication. It should be noted that, at present,
there is no well-known substitute for GGA-induced HSP22. As
aforementioned, certain cytokines or small molecule substances have
the capacity to trigger HSP22; nevertheless, there is little data
to confirm their safety and effectiveness. More optimal, secure and
efficient substitutes for GGA-induced HSP22 expression may be
discovered in the future as studies into the induction mechanism of
HSP22 continue and new drugs are developed.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82160085) and the Key Science and
Technology Innovation Projects of Jiangxi Provincial Health
Commission (grant no. 2024ZD007).
Availability of data and materials
Not applicable.
Authors' contributions
YC and YW were responsible for study conception and
design. YC and ML wrote the manuscript and were responsible for
making revisions. Data authentication is not applicable. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stetler RA, Gan Y, Zhang W, Liou AK, Gao
Y, Cao G and Chen J: Heat shock proteins: Cellular and molecular
mechanisms in the central nervous system. Prog Neurobiol.
92:184–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carra S, Alberti S, Arrigo PA, Benesch JL,
Benjamin IJ, Boelens W, Bartelt-Kirbach B, Brundel BJJM, Buchner J,
Bukau B, et al: The growing world of small heat shock proteins:
From structure to functions. Cell Stress Chaperones. 22:601–611.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yun CW, Kim HJ, Lim JH and Lee SH: Heat
shock proteins: Agents of cancer development and therapeutic
targets in anti-cancer therapy. Cells. 9:602019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benjamin IJ and McMillan DR: Stress (heat
shock) proteins: Molecular chaperones in cardiovascular biology and
disease. Circ Res. 83:117–132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Deniset JF and Pierce GN: Heat shock
proteins: Mediators of atherosclerotic development. Curr Drug
Targets. 16:816–826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nayak Rao S: The role of heat shock
proteins in kidney disease. J Transl Int Med. 4:114–117. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu Q, Metzler B, Jahangiri M and Mandal K:
Molecular chaperones and heat shock proteins in atherosclerosis. Am
J Physiol Heart Circ Physiol. 302:H506–H514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kappé G, Franck E, Verschuure P, Boelens
WC, Leunissen JA and de Jong WW: The human genome encodes 10
alpha-crystallin-related small heat shock proteins: HspB1-10. Cell
Stress Chaperones. 8:53–61. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kappé G, Verschuure P, Philipsen RL,
Staalduinen AA, Van de Boogaart P, Boelens WC and De Jong WW:
Characterization of two novel human small heat shock proteins:
Protein kinase-related HspB8 and testis-specific HspB9. Biochim
Biophys Acta. 1520:1–6. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morrow G, Hightower LE and Tanguay RM:
Small heat shock proteins: Big folding machines. Cell Stress
Chaperones. 20:207–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Haslbeck M, Franzmann T, Weinfurtner D and
Buchner J: Some like it hot: The structure and function of small
heat-shock proteins. Nat Struct Mol Biol. 12:842–846. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vos MJ, Hageman J, Carra S and Kampinga
HH: Structural and functional diversities between members of the
human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry.
47:7001–7011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smith CC, Yu YX, Kulka M and Aurelian L: A
novel human gene similar to the protein kinase (PK) coding domain
of the large subunit of herpes simplex virus type 2 ribonucleotide
reductase (ICP10) codes for a serine-threonine PK and is expressed
in melanoma cells. J Biol Chem. 275:25690–25699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benndorf R, Sun X, Gilmont RR, Biederman
KJ, Molloy MP, Goodmurphy CW, Cheng H, Andrews PC and Welsh MJ:
HSP22, a new member of the small heat shock protein superfamily,
interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J Biol
Chem. 276:26753–26761. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li XS, Xu Q, Fu XY and Luo WS: Heat shock
protein 22 overexpression is associated with the progression and
prognosis in gastric cancer. J Cancer Res Clin Oncol.
140:1305–1313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun X, Fontaine JM, Bartl I, Behnam B,
Welsh MJ and Benndorf R: Induction of Hsp22 (HspB8) by estrogen and
the metalloestrogen cadmium in estrogen receptor-positive breast
cancer cells. Cell Stress Chaperones. 12:307–319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki M, Matsushima-Nishiwaki R,
Kuroyanagi G, Suzuki N, Takamatsu R, Furui T, Yoshimi N, Kozawa O
and Morishige K: Regulation by heat shock protein 22 (HSPB8) of
transforming growth factor-α-induced ovary cancer cell migration.
Arch Biochem Biophys. 571:40–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsushima-Nishiwaki R, Toyoda H,
Takamatsu R, Yasuda E, Okuda S, Maeda A, Kaneoka Y, Yoshimi N,
Kumada T and Kozawa O: Heat shock protein 22 (HSPB8) reduces the
migration of hepatocellular carcinoma cells through the suppression
of the phosphoinositide 3-kinase (PI3K)/AKT pathway. Biochim
Biophys Acta Mol Basis Dis. 1863:1629–1639. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Modem S, Chinnakannu K, Bai U, Reddy GP
and Reddy TR: Hsp22 (HspB8/H11) knockdown induces Sam68 expression
and stimulates proliferation of glioblastoma cells. J Cell Physiol.
226:2747–2751. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang K, Yin W, Ma L, Liu Z and Li Q:
HSPB8 facilitates prostate cancer progression via activating the
JAK/STAT3 signaling pathway. Biochem Cell Biol. 101:1–11. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cristofani R, Piccolella M, Montagnani
Marelli M, Tedesco B, Poletti A and Moretti RM: HSPB8 counteracts
tumor activity of BRAF- and NRAS-mutant melanoma cells by
modulation of RAS-prenylation and autophagy. Cell Death Dis.
13:9732022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Ma S, Ye Z, Zheng Y, Zheng Z, Liu
X and Zhou X: Oncogenic DNA methyltransferase 1 activates the
PI3K/AKT/mTOR signalling by blocking the binding of HSPB8 and BAG3
in melanoma. Epigenetics. 18:22396072023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chowdary TK, Raman B, Ramakrishna T and
Rao CM: Mammalian Hsp22 is a heat-inducible small heat-shock
protein with chaperone-like activity. Biochem J. 381((Pt 2)):
379–387. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cristofani R, Rusmini P, Galbiati M,
Cicardi ME, Ferrari V, Tedesco B, Casarotto E, Chierichetti M,
Messi E, Piccolella M, et al: The regulation of the small heat
shock protein B8 in misfolding protein diseases causing
motoneuronal and muscle cell death. Front Neurosci. 13:7962019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rusmini P, Cristofani R, Galbiati M,
Cicardi ME, Meroni M, Ferrari V, Vezzoli G, Tedesco B, Messi E,
Piccolella M, et al: The role of the heat shock protein B8 (HSPB8)
in motoneuron diseases. Front Mol Neurosci. 10:1762017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bouhy D, Juneja M, Katona I, Holmgren A,
Asselbergh B, De Winter V, Hochepied T, Goossens S, Haigh JJ,
Libert C, et al: A knock-in/knock-out mouse model of
HSPB8-associated distal hereditary motor neuropathy and myopathy
reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol.
135:131–148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Depre C, Kim SJ, John AS, Huang Y, Rimoldi
OE, Pepper JR, Dreyfus GD, Gaussin V, Pennell DJ, Vatner DE, et al:
Program of cell survival underlying human and experimental
hibernating myocardium. Circ Res. 95:433–440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Depre C, Tomlinson JE, Kudej RK, Gaussin
V, Thompson E, Kim SJ, Vatner DE, Topper JN and Vatner SF: Gene
program for cardiac cell survival induced by transient ischemia in
conscious pigs. Proc Natl Acad Sci USA. 98:9336–9341. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hase M, Depre C, Vatner SF and Sadoshima
J: H11 has dose-dependent and dual hypertrophic and proapoptotic
functions in cardiac myocytes. Biochem J. 388:475–483. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Lizano P, Zhao X, Sui X, Dhar SK,
Shen YT, Vatner DE, Vatner SF and Depre C: Preemptive conditioning
of the swine heart by H11 kinase/Hsp22 provides cardiac protection
through inducible nitric oxide synthase. Am J Physiol Heart Circ
Physiol. 300:H1303–H1310. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sui X, Li D, Qiu H, Gaussin V and Depre C:
Activation of the bone morphogenetic protein receptor by
H11kinase/Hsp22 promotes cardiac cell growth and survival. Circ
Res. 104:887–895. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Laure L, Long R, Lizano P, Zini R,
Berdeaux A, Depre C and Morin D: Cardiac H11 kinase/Hsp22
stimulates oxidative phosphorylation and modulates mitochondrial
reactive oxygen species production: Involvement of a nitric
oxide-dependent mechanism. Free Radic Biol Med. 52:2168–2176. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Depre C, Wang L, Sui X, Qiu H, Hong C,
Hedhli N, Ginion A, Shah A, Pelat M, Bertrand L, et al: H11 kinase
prevents myocardial infarction by preemptive preconditioning of the
heart. Circ Res. 98:280–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qiu H, Lizano P, Laure L, Sui X, Rashed E,
Park JY, Hong C, Gao S, Holle E, Morin D, et al: H11 kinase/heat
shock protein 22 deletion impairs both nuclear and mitochondrial
functions of STAT3 and accelerates the transition into heart
failure on cardiac overload. Circulation. 124:406–415. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu W, Sun X, Shi X, Lai L, Wang C, Xie M,
Qin G and Qiu H: Hsp22 deficiency induces age-dependent cardiac
dilation and dysfunction by impairing autophagy, metabolism, and
oxidative response. Antioxidants (Basel). 10:15502021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gober MD, Smith CC, Ueda K, Toretsky JA
and Aurelian L: Forced expression of the H11 heat shock protein can
be regulated by DNA methylation and trigger apoptosis in human
cells. J Biol Chem. 278:37600–37609. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Verschuure P, Tatard C, Boelens WC,
Grongnet JF and David JC: Expression of small heat shock proteins
HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal
developing pig. Eur J Cell Biol. 82:523–530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Taylor RP and Benjamin IJ: Small heat
shock proteins: A new classification scheme in mammals. J Mol Cell
Cardiol. 38:433–444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Knowlton AA and Sun L: Heat-shock
factor-1, steroid hormones, and regulation of heat-shock protein
expression in the heart. Am J Physiol Heart Circ Physiol.
280:H455–H464. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eriksson M, Jokinen E, Sistonen L and
Leppä S: Heat shock factor 2 is activated during mouse heart
development. Int J Dev Biol. 44:471–477. 2000.PubMed/NCBI
|
|
41
|
Voellmy R: On mechanisms that control heat
shock transcription factor activity in metazoan cells. Cell Stress
Chaperones. 9:122–133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Morimoto RI: Proteotoxic stress and
inducible chaperone networks in neurodegenerative disease and
aging. Genes Dev. 22:1427–1438. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lizano P, Rashed E, Kang H, Dai H, Sui X,
Yan L, Qiu H and Depre C: The valosin-containing protein promotes
cardiac survival through the inducible isoform of nitric oxide
synthase. Cardiovasc Res. 99:685–693. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
D'Autréaux B and Toledano MB: ROS as
signalling molecules: Mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Prasad S, Gupta SC and Tyagi AK: Reactive
oxygen species (ROS) and cancer: Role of antioxidative
nutraceuticals. Cancer Lett. 387:95–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cheung EC and Vousden KH: The role of ROS
in tumour development and progression. Nat Rev Cancer. 22:280–297.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morin D, Long R, Panel M, Laure L, Taranu
A, Gueguen C, Pons S, Leoni V, Caccia C, Vatner SF, et al: Hsp22
overexpression induces myocardial hypertrophy, senescence and
reduced life span through enhanced oxidative stress. Free Radic
Biol Med. 137:194–200. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bou-Teen D, Kaludercic N, Weissman D,
Turan B, Maack C, Di Lisa F and Ruiz-Meana M: Mitochondrial ROS and
mitochondria-targeted antioxidants in the aged heart. Free Radic
Biol Med. 167:109–124. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yu Y, Hu LL, Liu L, Yu LL, Li JP, Rao JA,
Zhu LJ, Bao HH and Cheng XS: Hsp22 ameliorates
lipopolysaccharide-induced myocardial injury by inhibiting
inflammation, oxidative stress, and apoptosis. Bioengineered.
12:12544–12554. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Boulgakoff L, D'Amato G and Miquerol L:
Molecular regulation of cardiac conduction system development. Curr
Cardiol Rep. 26:943–952. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Miller DR and Thorburn A: Autophagy and
organelle homeostasis in cancer. Dev Cell. 56:906–918. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tedesco B, Vendredy L, Adriaenssens E,
Cozzi M, Asselbergh B, Crippa V, Cristofani R, Rusmini P, Ferrari
V, Casarotto E, et al: HSPB8 frameshift mutant aggregates weaken
chaperone-assisted selective autophagy in neuromyopathies.
Autophagy. 19:2217–2239. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shirakabe A, Ikeda Y, Sciarretta S,
Zablocki DK and Sadoshima J: Aging and autophagy in the heart. Circ
Res. 118:1563–1576. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dewanjee S, Vallamkondu J, Kalra RS, John
A, Reddy PH and Kandimalla R: Autophagy in the diabetic heart: A
potential pharmacotherapeutic target in diabetic cardiomyopathy.
Ageing Res Rev. 68:1013382021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rabinovich-Nikitin I, Kirshenbaum E and
Kirshenbaum LA: Autophagy, clock genes, and cardiovascular disease.
Can J Cardiol. 39:1772–1780. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Titus AS, Sung EA, Zablocki D and
Sadoshima J: Mitophagy for cardioprotection. Basic Res Cardiol.
118:422023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Arndt V, Dick N, Tawo R, Dreiseidler M,
Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et
al: Chaperone-assisted selective autophagy is essential for muscle
maintenance. Curr Biol. 20:143–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ulbricht A, Eppler FJ, Tapia VE, van der
Ven PF, Hampe N, Hersch N, Vakeel P, Stadel D, Haas A, Saftig P, et
al: Cellular mechanotransduction relies on tension-induced and
chaperone-assisted autophagy. Curr Biol. 23:430–435. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Carra S, Seguin SJ and Landry J: HspB8 and
Bag3: A new chaperone complex targeting misfolded proteins to
macroautophagy. Autophagy. 4:237–239. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liang Z, Zhang S, Zou Z, Li J, Wu R, Xia
L, Shi G, Cai J, Tang J and Jian J: Functional characterization of
BAG3 in orange-spotted grouper (Epinephelus coioides) during viral
infection. Fish Shellfish Immunol. 122:465–475. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Peng S, Yu Y, Li J, Jiang D, Xu G, Wu L
and Hu J: Hsp22 pretreatment protects against LPS-induced
hippocampal injury by alleviating neuroinflammation and apoptosis
by regulating the NLRP3/Caspase1/IL-1β signaling pathway in mice.
Aging (Albany NY). 15:1977–2004. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lan Y, Wang Y, Huang K and Zeng Q: Heat
shock protein 22 attenuates doxorubicin-induced cardiotoxicity via
regulating inflammation and apoptosis. Front Pharmacol. 11:2572020.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Depre C, Hase M, Gaussin V, Zajac A, Wang
L, Hittinger L, Ghaleh B, Yu X, Kudej RK, Wagner T, et al: H11
kinase is a novel mediator of myocardial hypertrophy in vivo. Circ
Res. 91:1007–1014. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rashed E, Lizano P, Dai H, Thomas A,
Suzuki CK, Depre C and Qiu H: Heat shock protein 22 (Hsp22)
regulates oxidative phosphorylation upon its mitochondrial
translocation with the inducible nitric oxide synthase in mammalian
heart. PLoS One. 10:e01195372015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bolli R: Cardioprotective function of
inducible nitric oxide synthase and role of nitric oxide in
myocardial ischemia and preconditioning: An overview of a decade of
research. J Mol Cell Cardiol. 33:1897–1918. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yamagami K, Yamamoto Y, Ishikawa Y,
Yonezawa K, Toyokuni S and Yamaoka Y: Effects of
geranyl-geranyl-acetone administration before heat shock
preconditioning for conferring tolerance against
ischemia-reperfusion injury in rat livers. J Lab Clin Med.
135:465–475. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ooie T, Kajimoto M, Takahashi N, Shinohara
T, Taniguchi Y, Kouno H, Wakisaka O, Yoshimatsu H and Saikawa T:
Effects of insulin resistance on geranylgeranylacetone-induced
expression of heat shock protein 72 and cardioprotection in
high-fat diet rats. Life Sci. 77:869–881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
He D, Song X and Li L:
Geranylgeranylacetone protects against cerebral ischemia and
reperfusion injury: HSP90 and eNOS phosphorylation involved. Brain
Res. 1599:150–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sysa-Shah P, Xu Y, Guo X, Pin S, Bedja D,
Bartock R, Tsao A, Hsieh A, Wolin MS, Moens A, et al:
Geranylgeranylacetone blocks doxorubicin-induced cardiac toxicity
and reduces cancer cell growth and invasion through RHO pathway
inhibition. Mol Cancer Ther. 13:1717–1728. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Brundel BJ, Henning RH, Ke L, van Gelder
IC, Crijns HJ and Kampinga HH: Heat shock protein upregulation
protects against pacing-induced myolysis in HL-1 atrial myocytes
and in human atrial fibrillation. J Mol Cell Cardiol. 41:555–562.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Brundel BJ, Shiroshita-Takeshita A, Qi X,
Yeh YH, Chartier D, van Gelder IC, Henning RH, Kampinga HH and
Nattel S: Induction of heat shock response protects the heart
against atrial fibrillation. Circ Res. 99:1394–1402. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sakabe M, Shiroshita-Takeshita A, Maguy A,
Brundel BJ, Fujiki A, Inoue H and Nattel S: Effects of a heat shock
protein inducer on the atrial fibrillation substrate caused by
acute atrial ischaemia. Cardiovasc Res. 78:63–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang D, Ke L, Mackovicova K, Van Der Want
JJ, Sibon OC, Tanguay RM, Morrow G, Henning RH, Kampinga HH and
Brundel BJ: Effects of different small HSPB members on contractile
dysfunction and structural changes in a Drosophila melanogaster
model for atrial fibrillation. J Mol Cell Cardiol. 51:381–389.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chang SL, Chen YC, Hsu CP, Kao YH, Lin YK,
Lai YJ, Yeh HI, Higa S, Chen SA and Chen YJ: Heat shock protein
inducer modifies arrhythmogenic substrate and inhibits atrial
fibrillation in the failing heart. Int J Cardiol. 168:4019–4026.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
van Marion DM, Hu X, Zhang D,
Hoogstra-Berends F, Seerden JG, Loen L, Heeres A, Steen H, Henning
RH and Brundel BJ: Screening of novel HSP-inducing compounds to
conserve cardiomyocyte function in experimental atrial
fibrillation. Drug Des Devel Ther. 13:345–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hu X, Li J, van Marion DMS, Zhang D and
Brundel BJJM: Heat shock protein inducer GGA*-59 reverses
contractile and structural remodeling via restoration of the
microtubule network in experimental atrial fibrillation. J Mol Cell
Cardiol. 134:86–97. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
van Marion DMS, Dorsch L, Hoogstra-Berends
F, Kakuchaya T, Bockeria L, de Groot NMS and Brundel BJJM: Oral
geranylgeranylacetone treatment increases heat shock protein
expression in human atrial tissue. Heart Rhythm. 17:115–122. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Waddingham MT, Sequeira V, Kuster DWD, Dal
Canto E, Handoko ML, de Man FS, da Silva Gonçalves Bós D,
Ottenheijm CA, Shen S, van der Pijl RJ, et al:
Geranylgeranylacetone reduces cardiomyocyte stiffness and
attenuates diastolic dysfunction in a rat model of cardiometabolic
syndrome. Physiol Rep. 11:e157882023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sanbe A, Daicho T, Mizutani R, Endo T,
Miyauchi N, Yamauchi J, Tanonaka K, Glabe C and Tanoue A:
Protective effect of geranylgeranylacetone via enhancement of HSPB8
induction in desmin-related cardiomyopathy. PLoS One. 4:e53512009.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Marunouchi T, Inomata S, Sanbe A, Takagi N
and Tanonaka K: Protective effect of geranylgeranylacetone via
enhanced induction of HSPB1 and HSPB8 in mitochondria of the
failing heart following myocardial infarction in rats. Eur J
Pharmacol. 730:140–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Gong R, Li XY, Chen HJ, Xu CC, Fang HY,
Xiang J and Wu YQ: Role of heat shock protein 22 in the protective
effect of geranylgeranylacetone in response to oxidized-LDL. Drug
Des Devel Ther. 13:2619–2632. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jiang B, Liu Y, Liang P, Li Y, Liu Z, Tong
Z, Lv Q, Liu M and Xiao X: MicroRNA-126a-5p enhances myocardial
ischemia-reperfusion injury through suppressing Hspb8 expression.
Oncotarget. 8:94172–94187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ren AJ, Chen C, Zhang S, Liu M, Wei C,
Wang K, Ma X, Song Y, Wang R, Zhang H, et al: Zbtb20 deficiency
causes cardiac contractile dysfunction in mice. FASEB J.
34:13862–13876. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Martin TG, Delligatti CE, Muntu NA,
Stachowski-Doll MJ and Kirk JA: Pharmacological inhibition of
BAG3-HSP70 with the proposed cancer therapeutic JG-98 is toxic for
cardiomyocytes. J Cell Biochem. 123:128–141. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cheng J, Ji M, Jing H and Lin H: DUSP12
ameliorates myocardial ischemia-reperfusion injury through
HSPB8-induced mitophagy. J Biochem Mol Toxicol. 37:e233102023.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Vieri M, Geng H, Patterson JB, Panse J,
Wilop S, Samali A, Chevet E and Kharabi Masouleh B: Deregulated
expression of the HSP40 family members Auxilin-1 and −2 is
indicative of proteostasis imbalance and predicts patient outcome
in Ph(+) leukemia. Exp Hematol Oncol. 5:52015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Criado-Marrero M, Gebru NT, Blazier DM,
Gould LA, Baker JD, Beaulieu-Abdelahad D and Blair LJ: Hsp90
co-chaperones, FKBP52 and Aha1, promote tau pathogenesis in aged
wild-type mice. Acta Neuropathol Commun. 9:652021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Shilova V, Zatsepina O, Zakluta A, Karpov
D, Chuvakova L, Garbuz D and Evgen'ev M: Age-dependent expression
profiles of two adaptogenic systems and thermotolerance in
Drosophila melanogaster. Cell Stress Chaperones. 25:305–315. 2020.
View Article : Google Scholar : PubMed/NCBI
|