Introduction
Low back pain (LBP) is one of the leading causes of
disability worldwide and myelopathy associated with Modic changes
(MCs) are considered highly specific for discogenic LBP. According
to the signal intensity in magnetic resonance imaging (MRI),
different types of MC can be classified (1). Modic type 1 changes (MC1) lesions are
characterized by low signal intensity on T1-weighted MRI and high
signal intensity on T2-weighted images. Conversely, Modic type 2
changes (MC2) lesions exhibit high signal intensity on T1-weighted
images and low signal intensity on T2-weighted images. MC3) lesions
demonstrate low signal intensity on both T1- and T2-weighted
images. These signal intensities are thought to reflect underlying
pathological processes: MC1 is typically associated with non-fatty
inflammatory changes, MC2 with fatty degeneration and MC3 with
sclerotic bone changes. Furthermore, these MC types are not static
and may transition from one type to another over time, suggesting a
dynamic interaction between the pathological progression of the
vertebral endplate and bone marrow degeneration (1,2).
Currently, numerous clinical studies have been
conducted to explore the pain association, prevalence and imaging
manifestations of MCs (3–6). Meanwhile, significant progress has
been made in understanding the pathological mechanisms of MC.
Latest investigations propose that fibrosis is likely a key
pathophysiological trait in MC1, while MC2 is thought to be linked
to fibroinflammatory changes near the endplate, possibly involving
the complement system (7,8). The present review aimed to synthesize
the current understanding of the molecular and cellular properties
inherent to MC, providing deeper insights into the disease
mechanisms that drive these manifestations. Revealing these
mechanisms will help elucidate the potential role of MC in LBP and
deepen our understanding of MC pathophysiology.
Occurrence and development of MCs
The pathophysiology of MCs is multifactorial,
including heavy physical load, autoimmune inflammation, endplate
injury, Cutibacterium acnes infection, genetic
susceptibility, diabetes and other metabolic conditions (9–13).
MC is strongly associated with intervertebral disc degeneration
(DD) and endplate rupture; however, the exact causal relationship
between these phenomena remains uncertain, with some studies
suggesting bidirectional effects (14). These factors contribute to
morphological changes in the intervertebral disc and surrounding
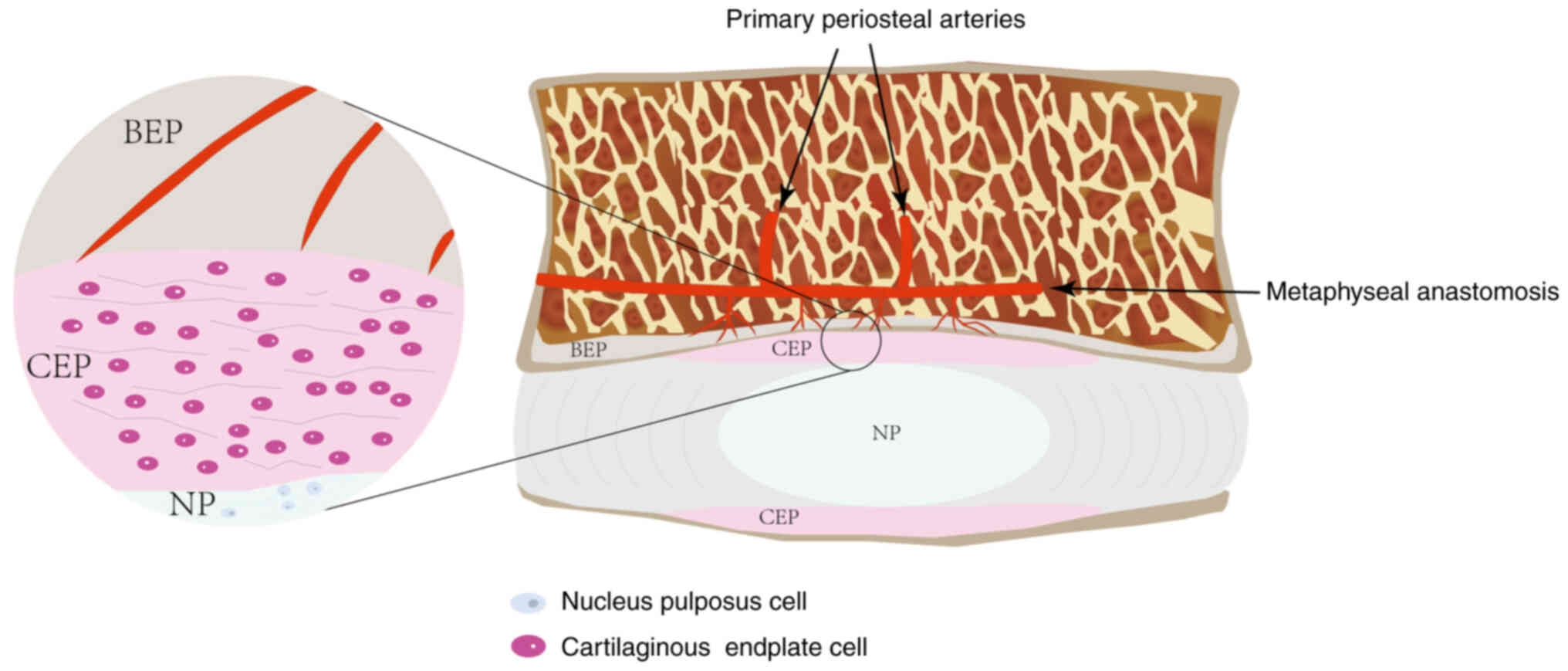
tissues. During embryonic development, nucleus pulposus (NP) cells
become isolated from the circulatory system, leading to
insufficient blood supply and an acidic environment that
facilitates immune isolation and the accumulation of metabolic
waste (15) (Fig. 1). When endplate damage occurs, NP
immunogens may be exposed, resulting in the infiltration of M1
macrophages and triggering immune-mediated inflammatory responses
(16). This destruction may also
create pathways between the bone marrow and intervertebral discs,
promoting osteoclast activity and impairing normal bone marrow
function through inflammatory signals from the intervertebral discs
(17–19). Additionally, microfractures of the
endplate can lead to increased expression of type II collagen,
catabolic enzymes and pro-inflammatory proteins, which in some
cases may exacerbate DD (20,21).
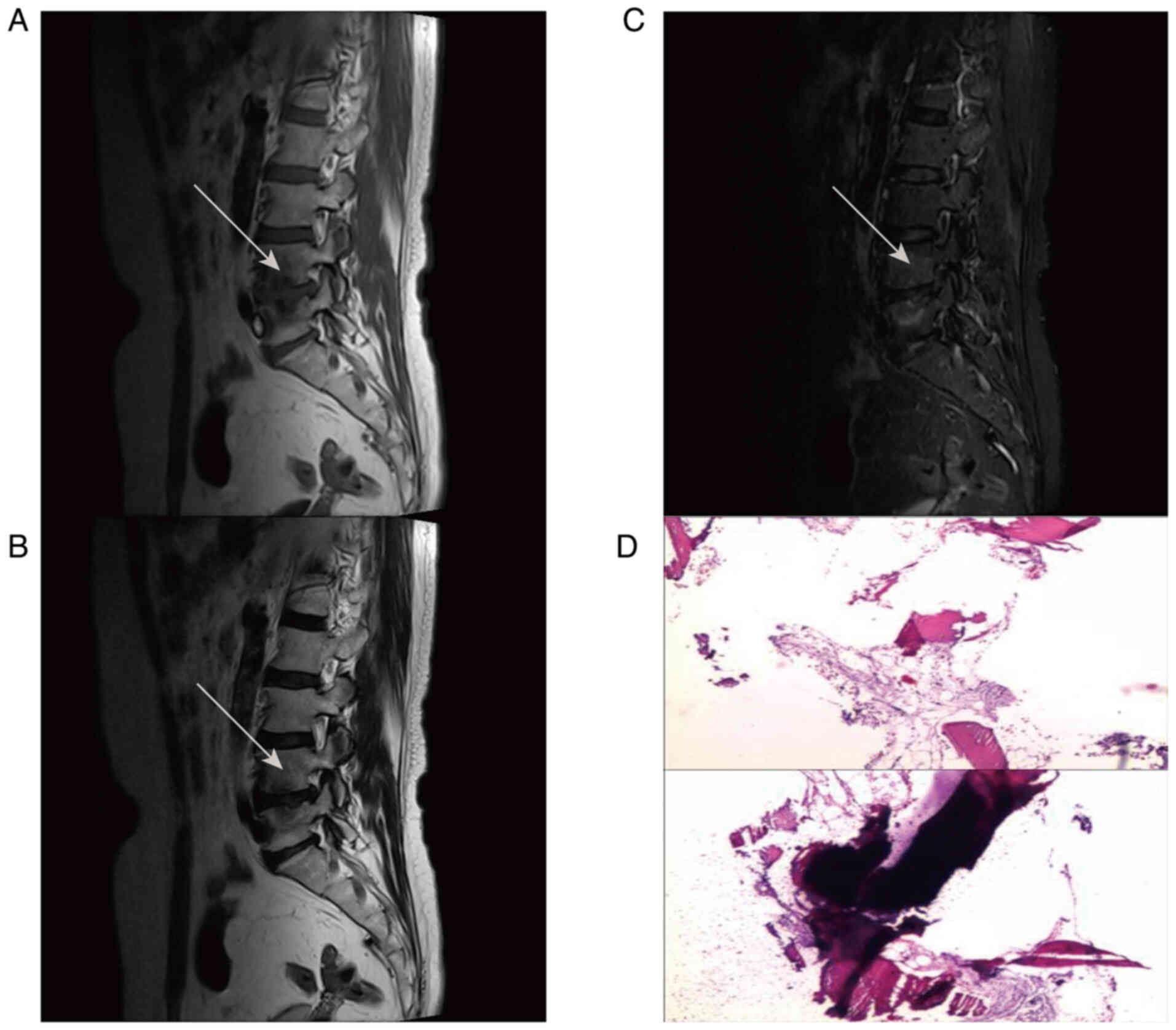
However, not all cases of MC necessarily lead to DD.
Some studies suggest that the degree of signal changes in vertebral
endplates may be associated with an increased likelihood of DD,
although the strength and potential mechanisms of this association
are still under investigation (20,22).
MC may exhibit high specificity (≥96%) in predicting DD, although
its low sensitivity suggests that DD does not always result in MC
(23).
Histological comparisons between MC1 and MC2 have
uncovered distinct tissue features and recent investigations have
broadened our understanding to include the molecular and cellular
dynamics within the bone marrow microenvironment. This has led to a
clearer picture of the pathophysiological variations between these
two types. Modic et al (1)
observed that bone marrow biopsies from both MC1 and MC2 lesions
are predominantly composed of fibrovascular granulation tissue.
They proposed the hypothesis that MC2 could be a progression stage
from MC1, characterized by an increased presence of adipocytes and
the conversion of red marrow to yellow (1). However, this progression is not
consistently linear, necessitating further research to clarify the
relationship. Perilli et al (4) noted significant bone loss and active
remodeling in MC1, whereas MC2 exhibited a decrease in bone
formation. Conversely, MC3 is linked to a seemingly stable phase of
bone remodeling, characterized by an increase in bone volume
fraction and trabecular thickness (4). The interaction between the bone
marrow in MC and the adjacent intervertebral discs, especially the
fibrotic and pro-inflammatory cross-talk, is hypothesized to be
pivotal in the pathophysiology of MC (24).
Studies have shed new light on the bone marrow
environment of MC1. Heggli et al (8) observed an upregulation of fibroblast
colony-forming units in MC1, an increase in levels of leptin
receptor (LEPR)-positive bone marrow stromal cells (BMSCs) and a
decrease in lipogenic differentiation ability. These findings
suggest that changes in the bone marrow microenvironment could
contribute to the fibrotic and inflammatory processes observed in
MC1. Similar to MC1, MC2 exhibits inflammatory infiltration in the
bone marrow; however, unlike MC1, this infiltration occurs without
the involvement of lymphocytes or neutrophils, indicating a
distinct inflammatory pathway in MC2 (25). This difference raises questions
about the exact immune mechanisms involved, with some studies
suggesting that pro-inflammatory adipokines may play a more
significant role (26–28). Further investigation is warranted
to elucidate cellular factors driving this inflammatory response.
Conversely, inflammation in MC2 may be driven by pro-inflammatory
adipokines derived from adipocytes, which could activate the
complement system as an upstream regulator of inflammation
(7). Over time, neutrophil markers
observed during the acute/subacute repair phase were significantly
downregulated, while the macrophage marker CD14 was found to be
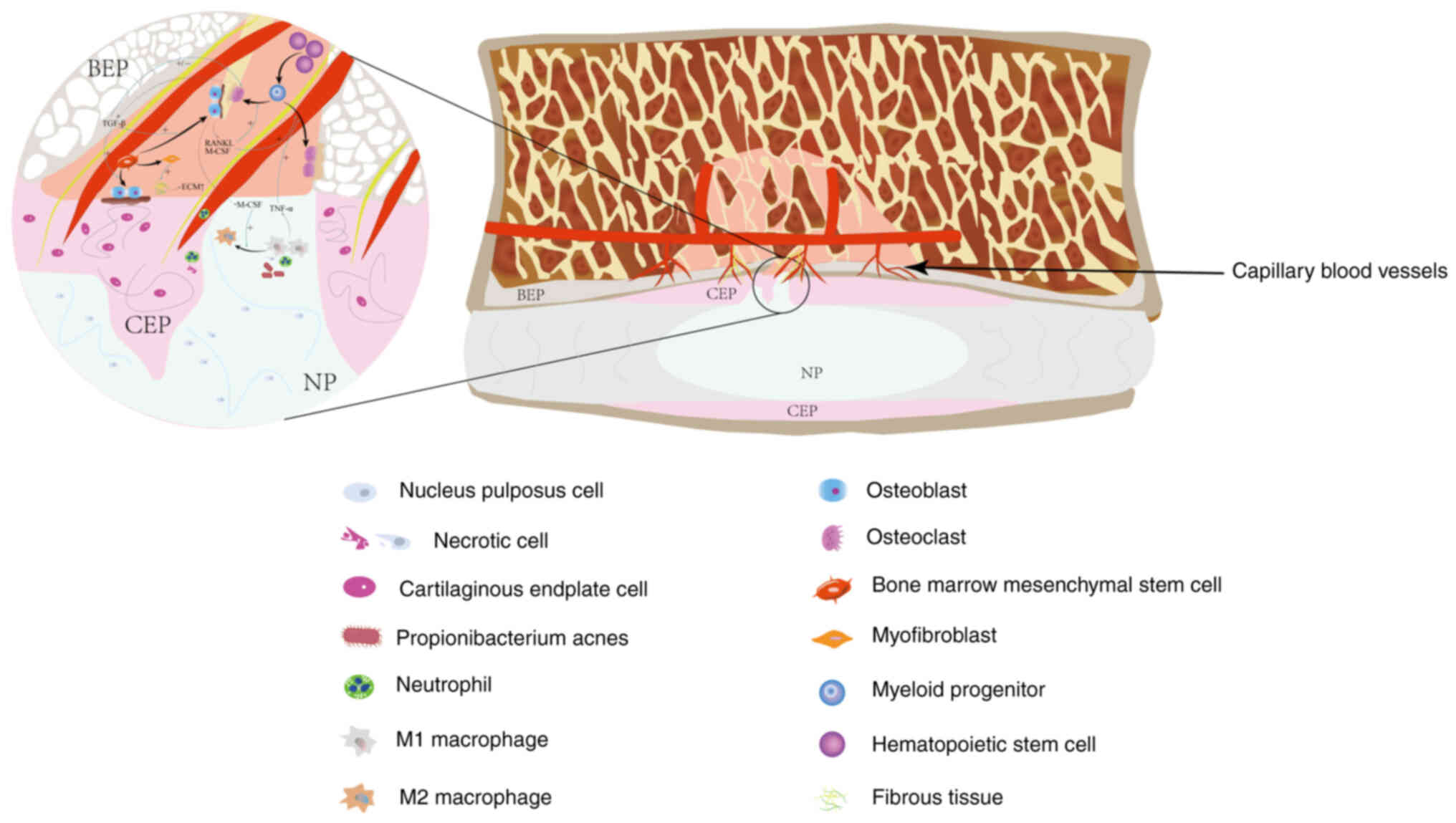
elevated in the bone marrow near the endplate (7). Based on these observations and
previous reports, the present review hypothesized that neutrophils
may be activated, matured and dysregulated in MC1, eventually being
cleared by macrophages in MC2 (29,30)
(Fig. 2).
Microfractures in the endplate may create pathways
between the intervertebral disc and bone marrow, potentially
leading to localized inflammation due to the presence of necrotic
cells, degraded extracellular matrix (ECM) components and bacterial
products. Some studies suggest that this may activate the
complement system, although the exact mechanism remains to be fully
elucidated (31–33). This activation may, in turn,
trigger the toll-like receptors (TLRs), although the direct
involvement of damage-associated molecular patterns (DAMPs) from
the ECM in MC still requires confirmation (34). Activation of the complement system
promotes the release of pro-inflammatory cytokines and may also
contribute to fibrosis and angiogenesis (7,35–37).
Although bone marrow lesions typically subside following acute
endplate injury, chronic stimulation can lead to persistent
inflammation and fibrosis, as seen in MC1 (38,39).
Heggli et al (7) found that
within MC2, the boundaries of endplate damage appeared to be
regressing, implying that chronic inflammation might cause these
defects to propagate towards the periphery. The mechanical load on
the edge of the endplate defect is higher and the involvement of
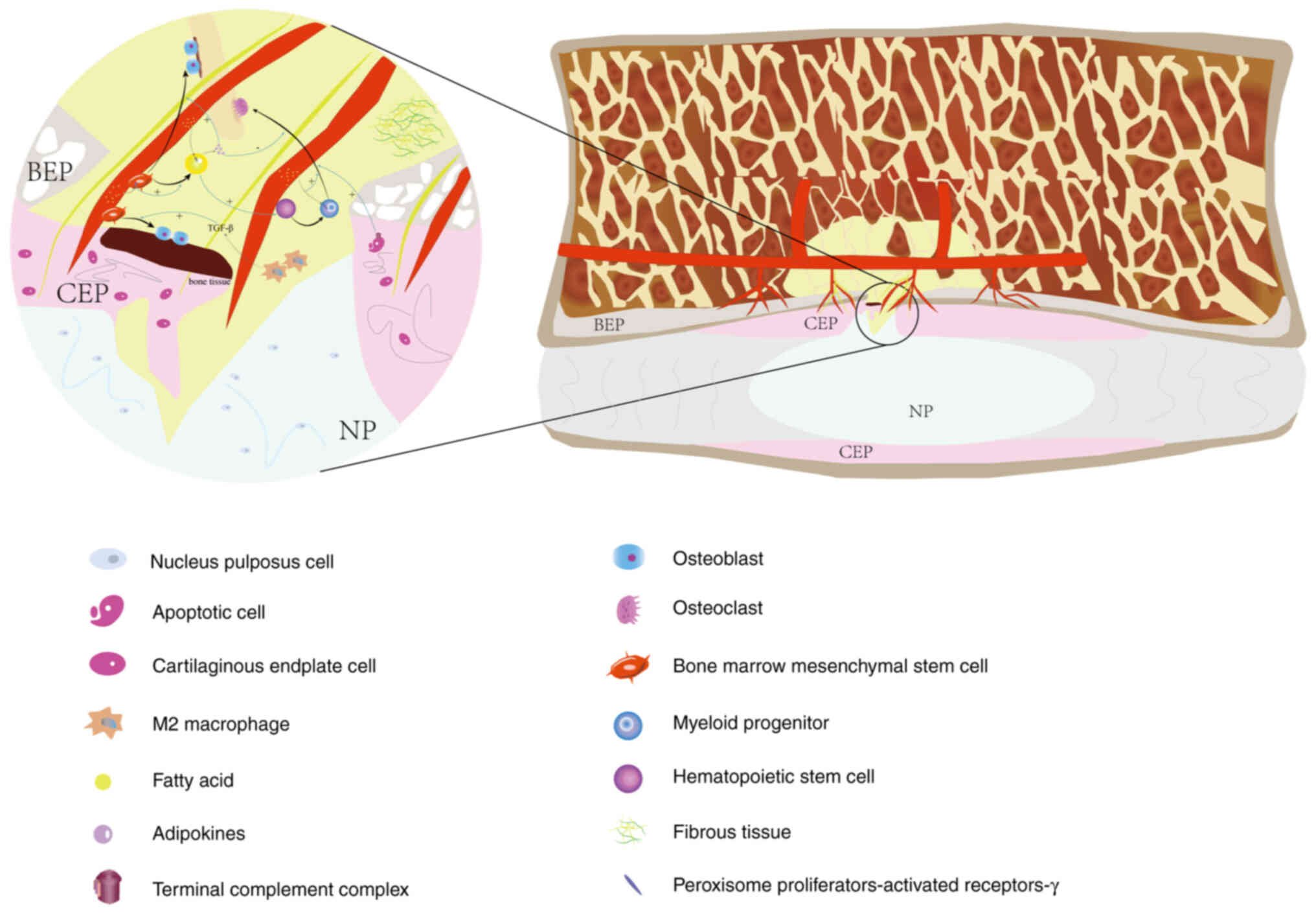
the complement system might drive this process (7,14).
Therefore, it was hypothesized that unresolved biomechanical
factors leading to MC could result in expanding endplate damage,
leading to a coexistence of acute inflammatory injury and chronic
repair in the affected region. This suggests that as endplate
damage worsens, complex dual repair reactions will occur (Figs. 3 and 4).
In MC1, BMSCs located in the perivascular region
exhibit a significant increase in C-X-C motif chemokine 12 (CXCL12)
and LEPR levels, promoting the formation of a hematopoietic niche
and enhancing osteoblast differentiation (8,40).
However, it must be noted that concurrent osteoclast activity may
inhibit the healing response in MC1 (33). Although LEPR+ CXCL12+ BMSCs have
the potential to differentiate into adipocytes, osteoblasts and
chondrocytes, their lipogenic differentiation may be impaired in
the pro-inflammatory environment of MC (24,41).
This impairment could trigger excessive fibrosis, as previous
studies have shown that BMSCs in MC1 exhibit a pro-fibrotic
phenotype (8). Decker et al
(40) proposed a thought-provoking
hypothesis that CXCL12+ LEPR+ cells may transdifferentiate into
myofibroblasts, contributing to the occurrence of
myelofibrosis.
In the later stage of MC development, there is a
clear shift in the bone marrow composition towards increased
adipogenesis. Elderly men, especially those with diabetes and lower
lumbar involvement, often exhibit higher adipose tissue content in
their spinal marrow (13,26). Long-term chronic inflammation,
coupled with the action of oxidized low-density lipoprotein, can
promote an increase in adipocyte hypertrophy and activate
peroxisome proliferator-activated receptor gamma (PPAR-γ), which
may indirectly inhibit bone marrow hematopoiesis (27,28,32,42,43).
This phenomenon could explain the observed decrease in neutrophil
counts during the MC2 stage. Although the effect of adipocytes on
bone marrow hematopoietic function may be multifaceted, their
positive role in supporting the survival of hematopoietic stem
cells cannot be denied (44,45).
However, during the MC3 stage, due to irreversible osteogenesis and
a decrease in adipocyte numbers, the overall regenerative potential
significantly diminishes (39,46).
In summary, the transition from MC1 to MC2/MC3 may
represent a continuous process from an acute inflammatory response
to more chronic reparative mechanisms, characterized by varying
inflammatory signatures and dynamic tissue remodeling. Future
studies aim at elucidating the molecular signals driving this
transformation could uncover novel therapeutic avenues,
particularly in modulating the inflammatory pathways and cellular
processes associated with these changes.
Involvement of the inflammatory system and
the complement system
Neutrophils
Neutrophils, also known as polymorphonuclear
leukocytes, are the initial responders to acute inflammation and
are rapidly recruited to the site of tissue injury due to their
critical role in the innate immune response (47). These cells originate from myeloid
precursors in the bone marrow, whose production is regulated by
granulocyte colony-stimulating factor (48). A recent study highlighted that the
presence of Cutibacterium acnes in MC1 significantly
influences the bone marrow microenvironment (49). In the presence of high
concentrations of Cutibacterium acnes, macrophages in the
bone marrow are exposed to pathogen-associated molecular patterns
following severe endplate damage, promoting the release of
macrophage colony-stimulating factor (M-CSF) and recruiting
monocytes and macrophages (50).
Meanwhile, TLR activation plays a crucial role in the recruitment
of neutrophils to the site of injury (50).
Neutrophils are crucial in early immune responses,
as they phagocytose bacteria, clear dead cells and eliminate
microbial debris (51). In the
context of MC, there is evidence suggesting that neutrophils
promote inflammation by forming neutrophil extracellular traps,
which may further activate the complement system and exacerbate
inflammatory responses (52,53).
Neutrophils also release matrix metalloproteinase-9 (MMP-9),
involved in the degradation of the ECM in NP cells, which may
exacerbate tissue damage and promote additional complement
activation (31). MMP-9 also plays
a crucial role in vascular regeneration, as it activates vascular
endothelial growth factor (VEGF) and promotes vascular growth at
the site of injury (54). Notably,
in MC1 patients with high levels of Cutibacterium acnes
GCNs, neutrophil degranulation was identified as a key enrichment
process, supporting a sustained antimicrobial response
characterized by the upregulation of cytokines such as IL-8 and
ENA-78, which attract and activate neutrophils (49).
By contrast, in individuals with lower
concentrations of Cutibacterium acnes, different immune
responses may predominate. The increased expression of IL-13 and
the activation of T and B cells suggest that an autoimmune
mechanism may also be involved in MC1 (24). Following severe endplate damage,
neutrophils accumulate and mature and may eventually be cleared by
macrophages, which helps to eliminate inflammation (49,55).
These findings suggest that the concentration of Cutibacterium
acnes may significantly affect the behavior of neutrophils,
ultimately impacting the inflammation and healing processes in
MC.
Macrophages
Macrophages originate from the mononuclear phagocyte
system and serve a critical role in maintaining tissue homeostasis
in conjunction with parenchymal cells (56). These cells can be roughly divided
into two phenotypes: M1 and M2 macrophages, each with distinct
immune functions. M1 macrophages are characterized by their
pro-inflammatory effects, secreting cytokines such as IL-1, IL-6
and tumor necrosis factor-α, actively participating in immune
surveillance and antigen presentation. By contrast, M2 macrophages
exhibit anti-inflammatory and immunosuppressive effects, releasing
cytokines such as IL-10 and transforming growth factor-β (TGF-β),
facilitating tissue repair and inflammation resolution (57). In the context of disc pathology,
particularly for NP cells that typically exist in an
immune-privileged environment, macrophage infiltration following EP
injury may disrupt this immune isolation. Once the immune barrier
is compromised, macrophages are hypothesized to cause ECM
degradation, potentially exacerbating inflammatory pain (58).
Although direct experimental evidence confirming the
role of macrophages in NP pathology is currently lacking, several
studies have proposed their involvement based on general
immunological mechanisms (8,39).
Studies have demonstrated that when immune protection is
compromised, macrophages may recognize NP cells and these damages
may worsen following EP damage (24,59).
Following EP injury, NP cells are exposed and it is hypothesized
that macrophages are recruited to the injury site in response to
chemotactic signals. Degenerated discs may further exacerbate
inflammation by releasing additional pro-inflammatory factors,
potentially promoting capillary infiltration and further
recruitment of macrophages (58).
As the condition progresses, M1 macrophages, which dominate the
initial inflammatory responses, are likely to be gradually replaced
by M2 macrophages.
The interaction between M1 and M2 macrophages is
hypothesized to have a profound effect on BMSCs and their
osteogenic differentiation process (60,61).
Specifically, M1 macrophages are thought to promote early
osteogenic differentiation, although they may not be as effective
in supporting complete mineralization of the bone matrix (62,63).
In practical observations, it was found that osteoblasts secreted
M-CSF and RANKL, which played key roles in promoting osteoclast
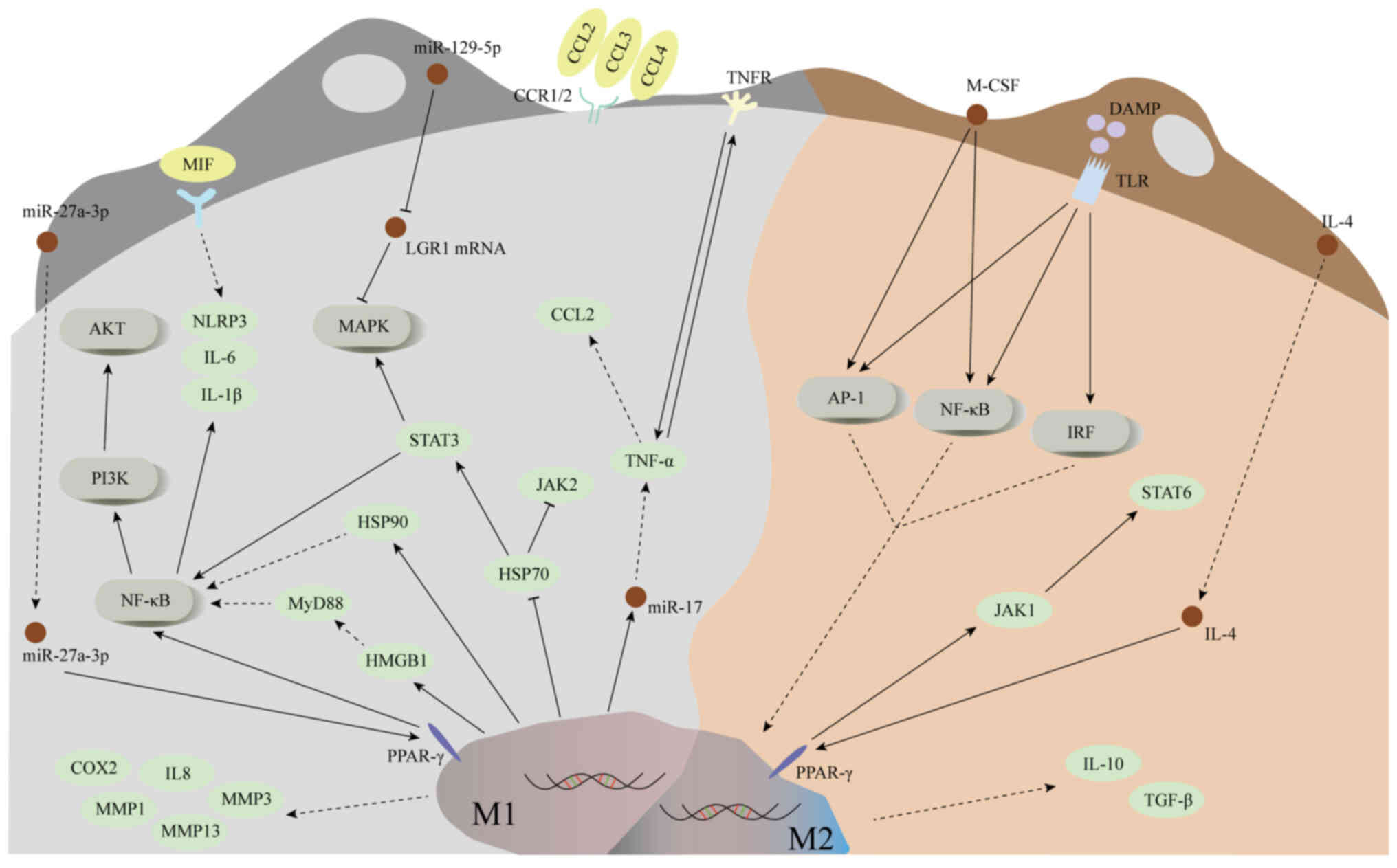
differentiation (64) (Fig. 5). Additionally, M-CSF can promote
the transformation of M1 macrophages to M2 macrophages by
upregulating the expression of c-Jun N-terminal kinase and nuclear
factor kappa-B (NF-κB) (65,66).
Conversely, M2 macrophages are hypothesized to
contribute to bone matrix mineralization and BMSC osteogenic
differentiation, thus aiding in tissue repair (63). The increase in the ratio of
osteoclasts to osteoblasts is thought to exacerbate the porous
structure in the EP, promote sensory nerve innervation and
potentially increase pain (64,67,68).
Overall, the coordinated regulation of M1 and M2 macrophages is
hypothesized to significantly affect neural sensitivity and
facilitate the repair of the EP at the site of injury. Future
research should focus on validating these mechanisms and their
clinical relevance (16).
M1 macrophages
M1 macrophages secrete TNF-α, a potent
pro-inflammatory cytokine, which plays a critical role in
regulating inflammation, lipid metabolism and apoptosis (69,70).
In vitro study has revealed that TNF-α may act as an inducer
of DD, mimicking its pathological mechanism (71). When immune barrier function of NP
is impaired, extensive infiltration of macrophages follows
(24). TNF-α secreted by M1
macrophages is hypothesized to activate NP cells, promoting their
expression of various chemokines such as chemoattractant cytokine
ligand (CCL2) and CCL3 (72).
Studies have shown that CCL2 is primarily responsible for
macrophage recruitment, while the CCL3-chemokine receptor 1 (CCR1)
signaling axis plays a crucial role in macrophage infiltration
(72,73). In vitro experiments have
shown that inhibiting CCR1 can effectively reduce the expression of
chronic inflammation in intervertebral discs and limit macrophage
migration (74). Additionally,
TNF-α enhances the expression of CCL4 by activating the p38-MAPK
and NF-κB signaling pathways in NP cells, thereby further promoting
macrophage infiltration (75). It
is known that TNF-α and IL-1β can upregulate MMPs in M1
macrophages, which contribute to ECM degradation (76). Moreover, these cytokines are also
hypothesized to stimulate the production of nerve growth factors,
which may lead to nerve infiltration and trigger LBP (77). It is worth noting that TNF-α has
also been found to induce the aging of NP cells, which may be
related to the decrease in intervertebral height and play a certain
role in the pathological development of MC.
IL-1β is considered a key inflammatory mediator
involved in numerous inflammatory processes. In vitro
co-culture studies with bone marrow and bone marrow monocytes in MC
models report significant upregulation of IL-1β, which promoted
additional pro-inflammatory cytokines, thereby amplifying the
inflammatory response (78) IL-1β
is associated with increased disc inflammation by upregulating
inflammatory mediators such as IL-6, IL-8 and prostaglandin E2
(PGE2), which may also lead to mitochondrial dysfunction and
accumulation of reactive oxygen species (79,80).
IL-1β also triggers apoptosis of NP cells through the NF-κB and
MAPK pathways, further exacerbating DD (81,82).
Inhibiting the expression of high mobility group box-1 protein and
blocking the activation of the MyD88/NF-κB pathway, as well as the
NLRP3 inflammasomes in NP cells, reduces the secretion of
inflammatory factors, prevents M1 macrophage polarization and
alleviates NP cell damage (83). A
study conducted by Zhang et al (84) demonstrated that the heat shock
protein 90 inhibitor, 17-AAG, attenuated the pro-inflammatory
activity of M1 macrophages by targeting the NF-κB and MAPK
pathways. Additionally, 17-AAG inhibited M1-induced NP cell
inflammation and catabolism in NP cells by enhancing the expression
of the HSP70, JAK2 and STAT3 pathways. Furthermore, 17-AAG
diminished the fibrotic phenotypes induced by macrophages in NP
cells by suppressing the expression of migration-inducing proteins
and reducing pathological angiogenesis (85).
IL-1α is released by monocyte macrophages and is
activated following apoptosis of necrotic cells, making it another
key inflammatory mediator (86).
Research has shown that co-culturing bone marrow mononuclear cells
with NP cells results in increased secretion of IL-10, which is
modulated by IL-1α (78). Necrotic
apoptosis regulates the maturation and release of IL-1α through the
activation of calpain (86). It is
hypothesized that IL-1α may accelerate DD by inhibiting ECM
synthesis and promoting ECM degradation. Additionally, IL-1α may
induce LBP by stimulating NP cells to release PGE2 and other
inflammatory mediators, which may activate nerve roots infiltrating
the cartilaginous endplate (87).
Furthermore, the upstream regulatory mechanisms of
inflammatory cytokines have been extensively investigated. For
instance, transfection of M1 macrophages with miR-17 significantly
increases the release of TNF-α, indicating a correlation between
miR-17 and increased TNF-α secretion (88). Moreover, it has been reported that
extracellular vesicles from BMSCs can deliver miR-129-5p to NP
cells, thereby downregulating the p38-MAPK signaling pathway.
miR-129-5p can also bind to the 3′-untranslated region of
leucine-rich glycoprotein-1 (LRG1), inhibiting its expression,
which may inhibit M1 macrophage polarization and foster M2
macrophage polarization along with the release of anti-inflammatory
factors (89) (Fig. 5). Furthermore, exosomes from
myeloid cells containing miR-27a-3p have been shown to target the
PPARγ/NFκB/PI3K/AKT signaling pathway, thereby promoting the
polarization of M1 macrophages (90).
M2 macrophages
M2 macrophage polarization is regulated by cytokines
and microbial byproducts in tissues (91). With prolonged immune exposure to
microfractures of the endplate, the expression of IL-1β is
hypothesized to decrease, while the concentration of IL-4 may
gradually increase. This increase is thought to promote the
polarization of M1 to M2 macrophages at the site of injury
(91,92). During the process of clearing
apoptotic neutrophils, monocyte-derived macrophages have been
reported to secrete IL-10, utilizing the STAT signaling pathway
(93). In a model of inflammatory
bowel disease, IL-10 was shown to inhibit mTORC1 activation through
the STAT3 pathway, thereby potentially modulating macrophage
metabolic activity and promoting an anti-inflammatory phenotype
(94). Additionally, activation of
the PPAR-γ receptor has been proposed to promote M2 polarization
through its interaction with the JAK1-STAT6 pathway (95,96).
Despite the partial understanding of the precise anti-inflammatory
mechanisms of IL-10, M2 macrophages are hypothesized to be crucial
in tissue repair by releasing TGF-β, particularly in granulation
tissue rich in M2 macrophages (97). The downregulation of miR-133a is
associated with the loss of type II collagen targeting MMP9 during
DD, supporting the pathological role of MMP9 (98). Additionally, MMP9 is hypothesized
to activate latent TGF-β by inducing fibroblast contraction,
providing insights into the potential role of TGF-β in disc
degeneration-related fibrosis (99). TGF-β is a key regulatory factor in
fibrosis, promoting the differentiation of BMSCs into
myofibroblasts, a hallmark of MC (100). Although data on TGF-β activation
factors are still limited, MMP9-mediated TGF-β stimulation, as
demonstrated in lung fibroblasts, suggests its possible involvement
in MC fibrosis, indicating that MMP9 can trigger fibrosis
activation (8,99). TGF-β acts through the Smad
signaling pathway, promoting the differentiation of BMSCs into
osteoblasts and regulating the fibrotic microenvironment (101). However, in the context of MC,
this mechanism of promoting fibrosis is still speculative and
requires further investigation (102,103).
BMSC
Myelofibrosis is a potential adverse effect within
the pathophysiological spectrum of MC1 (37). Decker et al (40) confirmed that CXCL12+ LEPR+ BMSCs
are the predominant source of myofibroblasts in primary
myelofibrosis. Additionally, in comparison with MC2, CD90+ BMSCs
are observed more frequently in the fibrotic microenvironment of
MC1 (25). In MC1, the three
critical signaling pathways of Notch, Wnt/β-catenin and Hedgehog
are prominently enriched in perivascular BMSCs, thereby playing a
pivotal role in driving the pro-fibrotic phenotype (8). The Hedgehog and Wnt/β-catenin
signaling pathways facilitate osteoblast differentiation while
inhibiting adipocyte differentiation (104,105). However, the Notch pathway
exhibits a dual role: It is essential for osteogenesis but can also
inhibit osteogenesis under specific conditions. For example,
studies indicate that the introduction of Notch ligands or
overexpression of the Notch target gene hairy and enhancer of split
1 in MSCs can suppress the expression of PPARγ and C/EBPα, thereby
inhibiting lipogenesis and promoting osteogenesis; conversely,
Notch can also inhibit osteogenesis by suppressing the
Wnt/β-catenin pathway (106).
Consequently, the inhibition of lipid differentiation observed in
MC1 may result from the interaction of these signaling pathways.
Nonetheless, the precise mechanism driving the differentiation of
LEPR-high BMSCs into myofibroblasts remains elusive and may be
initiated by the ‘thwarted healing response’ hypothesis proposed by
Dudli et al (37).
While direct research on adipocyte-predisposed
differentiation of BMSCs in MC2 pathology is lacking, Dudli et
al (37) used osteoarthritis
as a model to speculate on the possible mechanisms involved in MC2.
They hypothesized that TLRs played a pivotal role in both
osteoarthritis and MC pathophysiology (37). In osteoarthritis, TLRs act as
receptors for DAMPs generated by ECM degradation. The ECM
degradation associated with MC-induced DD may chronically activate
TLRs, prompting BMSCs to differentiate into adipocytes (107). As mentioned previously, these
adipocytes could release fatty acids, which might accelerate the
progression of MC through the PPARγ pathway.
Complement system
New evidence suggests that the complement system may
play a critical role in the pathology of MC, although specific
studies are still scarce. Heggli et al found that during the
progression of MC, the levels of complement proteins C5, C8 alpha
chain and factor B were elevated in the bone marrow (107), indicating activation of both
classical and alternative complement pathways (7,39).
It is hypothesized that mechanisms such as cell
death, matrix degradation and bacterial infection may trigger
complement activation in MC (107). Similar to osteoarthritis,
necrotic cells in MC may release DAMPs, which may activate the
classical complement pathway, based on reasonable inference of
similar conditions (34,39,108). As shown in the study by Dudli
et al (20), the presence
of necrotic cells in MC is supported by elevated levels of lactate
dehydrogenase. Additionally, degradation of the disc matrix may
release DAMPs, which may interact with the bone marrow through
ruptured endplates and further activate the complement system
(24). As proposed by Albert et
al (109), the presence of
Cutibacterium acnes in intervertebral discs exacerbated
these ruptures and increased the likelihood of complement
activation.
In osteoarthritis, the terminal complement complex
(TCC) is crucial in mediating cell death and inducing phenotypic
changes in chondrocytes following cartilage injury (110). Riegger et al (110) hypothesized that TCC could also
influence the cell death and healing dynamics of MC, potentially
creating a feedback loop that intensifies the condition.
Furthermore, complement fragments C3a and C5a, integral components
of TCC, exhibit notable pro-inflammatory effects, enhancing the
inflammatory response of osteoblasts and promoting osteoclast
formation, which may in turn affect the MCs (36,37).
The complement system is associated with chronic
inflammation involving MC. Specifically, C3a and C5a fragments can
stimulate the production of IL-1β, a critical pro-inflammatory
cytokine and VEGF, which is pivotal in supporting
neovascularization (35,36). While the formation of VEGF driven
by the complement system has been established in osteoarthritis, it
is reasonable to hypothesize that a similar mechanism operates in
MC (35). Furthermore, as
highlighted by Llorian-Salvador et al (34), complement activation is correlated
with elevated levels of TGF-β, a key factor in fibrotic
transformation.
The complement-mediated process may also be involved
in neurogenesis, which may explain the observed inward growth of
nerve fibers into ruptured endplates in MCs, a concept explored by
Fatoba et al (111). In
conclusion, although the role of the complement system in MC
remains largely speculative and current research is devoid of
direct evidence, it is widely hypothesized that complement
activation could trigger inflammation, angiogenesis, fibrosis and
neurogenesis, which may collectively drive the pathological
progression of MC. Further investigation is warranted to clarify
these mechanisms and their clinical implications.
Clinical implications
The current classification systems for MC, namely
MC1, MC2 and MC3, are generally deemed inadequate to fully
encompass the complexity of the pathological processes involved.
This classification scheme relies on key features such as endplate
morphology, bone marrow edema and the proportion of edematous-fatty
tissue. However, this simplification limits its ability to
effectively guide clinical decision-making (1). The MC classification is grounded in
key characteristics such as endplate morphology, extent of bone
marrow edema and the ratio of edematous-fatty tissue (Table I). Nevertheless, categorizing MC
into three types fails to capture the full complexity of the
condition, nor does it adequately assist in guiding clinical
decision-making for conditions such as discogenic LBP.
 | Table I.MC Classification. |
Table I.
MC Classification.
| First author/s,
year | Sample size | Results | (Refs.) |
|---|
| Xu et al,
2016 | 276 | MC can be further
subdivided into 10 distinct types: type I, type II, type III, type
I–II, type II–I, type I–III, type III–I, type II–III, type III–II
and type I–II-III. Endplate edema is responsible for most mixed
types, especially types I–II and II–I. It is important to note that
endplate sclerosis observed on CT images may not exclusively
indicate Modic type III but is actually present across all types of
MC. | (113) |
| Feng et al,
2018 | 133 | Three types of
endplate defects, focal, angular and erosive, were categorized.
These three types of endplate defects observed on conventional MR
images likely represent different underlying pathologies and may
significantly contribute to the pathogenesis of MC. | (126) |
| Udby et al,
2022 | - | According to the
area of MC in the sagittal section, the categories are defined as
follows: Type A corresponds to an MC area smaller than 25% of the
vertebral height, Type B encompasses an MC area between 25 and 50%
of the vertebral height and Type C refers to an MC area exceeding
50% of the vertebral height. | (127) |
| Rajasekaran et
al, 2024 | 1,530 | The DEBC was
established by incorporating STIR images and examining the entire
interdependent complex, categorizing conditions into: Type A: acute
inflammation; Type B: chronic persistence; Type C: latent; Type D:
inactive. Type B exhibits the highest incidence. Notably, the
highest ratio of cases requiring surgery was observed when both
disc herniation and DEBC type changes were present
concurrently. | (116) |
Studies have demonstrated an association between MC
and discogenic LBP, which directly influences patients' quality of
life (2,112). There remain challenges in
distinguishing MC from other spinal ailments, such as infections
and tumors, on MRI scans, particularly in instances of mixed MC, a
common clinical scenario. The persistent moderate inter-observer
variability further complicates this diagnostic challenge,
highlighting the ongoing uncertainty in MC diagnosis (113). Moreover, the moderate differences
among observers underscore a significant degree of uncertainty in
MC diagnosis (3).
The advancement of deep learning (DL) technology has
significantly enhanced medical imaging, enabling the automatic
extraction and classification of features from large datasets.
These advances have improved the diagnostic consistency of
radiologists in identifying MCs on MRI (114,115). This innovation is expected to
increase the diagnostic accuracy of spinal disorders.
While DL models substantially enhance MC detection,
they often neglect pathological processes occurring in adjacent
discs and tissues. To address this gap, Rajasekaran et al
(116) integrated disc herniation
and short tau inversion recovery (STIR) sequence data into MC
classification, thereby improving the predictive capability for
surgical intervention and postoperative complications. This study
did not comprehensively address the broader pathological changes
associated with MC. To enhance the understanding and management of
LBP, a more holistic approach is required, encompassing not only MC
and DD but also high-intensity zones, endplate defects and
vertebral bone marrow steatosis. Collecting detailed clinical data,
exploiting MRI sequences such as T1, T2 and STIR, as well as
employing advanced AI and scoring systems, such as the Japanese
Orthopaedic Association score, can contribute to the development of
predictive models. These models will enhance the precision of MC
staging and prognostic assessments, optimize clinical
decision-making and improve patient outcomes.
Furthermore, MC are frequently identified as a
factor associated with LBP and have been established as an
independent risk factor for severe and disabling episodes of LBP
(117,118). One study reported that the
prevalence of any form of MC was 43% in patients with nonspecific
LBP and/or sciatica, compared with just 6% in the general
population (119). The current
treatment options primarily include conservative management,
discectomy, lumbar fusion and intraosseous basivertebral nerve
radiofrequency neurotomy.
However, different treatment modalities for various
types of MC may lead to distinct outcomes. For instance, following
discectomy, the difference in improvement of LBP or Oswestry
disability index (ODI), or visual analogue scale (VAS) between
patients with or without preoperative MC did not exceed the minimal
clinically important difference (117). By contrast, a 2022 meta-analysis
concluded that the presence of preoperative MC did not
significantly influence clinical outcomes following lumbar
discectomy (120). However, this
meta-analysis did not compare improvements across different Modic
types at specific follow-up time points. A 2023 meta-analysis
further addressed this issue, revealing that patients with MC1
reported more LBP than those with other Modic types. Most of these
patients also reported greater leg pain than back pain.
Additionally, in patients with lumbar disc herniation and
preoperative MC1, functional improvements at the 2-year follow-up
were statistically lower compared with patients without MC1 or with
MC2 (121). As to intraosseous
basivertebral nerve radiofrequency neurotomy, a relatively recent
treatment, a 2022 meta-analysis reported success rates of 65 and
64% for pain relief at 6 and 12 months, respectively. The
improvement rates in ODI scores of ≥15 points at 6 and 12 months
were 75 and 75%, respectively. This indicates moderate-quality
evidence supporting the efficacy of intraosseous basivertebral
nerve radiofrequency neurotomy in alleviating pain and disability
in most patients with vertebrogenic LBP (122).
As previously discussed, bone marrow inflammation,
the immune response around the endplate and the higher density of
sensory nerve fibers in MC1 compared with other Modic types all
contribute to a relatively poorer long-term prognosis when the
surgical intervention does not involve the vertebral body and
endplate (4,24,123). Given the substantial overlap in
pain referral patterns from other spinal structures, including the
intervertebral disc, sacroiliac joints and facet joints, this may
also explain why the pain relief outcomes with intraosseous
basivertebral nerve radiofrequency neurotomy are not as favorable
as expected (124,125).
Conclusion
The multifaceted pathology of MC involves intricate
interactions between immune responses, chronic inflammation and
structural damage to the vertebral endplates. Key factors such as
endplate microfractures and Cutibacterium acnes infection
are still being elucidated, with the complement system being
pivotal in sustaining inflammation and fibrosis. Therapeutic
targets such as the RANK/RANKL/OPG axis and the Wnt/β-catenin
pathway hold promise for restoring endplate integrity and halting
the progression of MC.
The integration of AI into imaging techniques has
notably enhanced the diagnostic precision of MC, particularly in
identifying mixed types and assessing their effects on adjacent
tissues. Future research should focus on elucidating the molecular
mechanisms driving the advancement of MC, aiming to formulate more
effective and tailored therapies for discogenic LBP.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors participated in the study design and
manuscript writing. WJZ and SRZ were responsible for literature
collection, analysis and interpretation, contributing significantly
to the writing of the present review. JMZ and ZHX drafted the main
manuscript text and prepared the figures. ZY, WX and PL critically
reviewed the article for important intellectual content. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study received approval from the Ethics
Committee of Tongji Hospital Affiliated to Tongji Medical College,
Huazhong University of Science and Technology (approval no.
TJ-IRB202402127).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Modic MT, Steinberg PM, Ross JS, Masaryk
TJ and Carter JR: Degenerative disk disease: Assessment of changes
in vertebral body marrow with MR imaging. Radiology. 166((1 Pt 1)):
193–199. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thompson KJ, Dagher AP, Eckel TS, Clark M
and Reinig JW: Modic changes on MR images as studied with
provocative diskography: Clinical relevance-a retrospective study
of 2457 disks. Radiology. 250:849–855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carrino JA, Lurie JD, Tosteson AN,
Tosteson TD, Carragee EJ, Kaiser J, Grove MR, Blood E, Pearson LH,
Weinstein JN and Herzog R: Lumbar spine: Reliability of MR imaging
findings. Radiology. 250:161–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perilli E, Parkinson IH, Truong LH, Chong
KC, Fazzalari NL and Osti OL: Modic (endplate) changes in the
lumbar spine: Bone micro-architecture and remodelling. Eur Spine J.
24:1926–1934. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tarukado K, Ono T, Tono O, Tanaka H, Ikuta
K, Harimaya K and Doi T: Does modic change progresss with age?
Spine (Phila Pa 1976). 42:1805–1809. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brinjikji W, Diehn FE, Jarvik JG, Carr CM,
Kallmes DF, Murad MH and Luetmer PH: MRI findings of disc
degeneration are more prevalent in adults with low back pain than
in asymptomatic controls: A systematic review and meta-analysis.
AJNR Am J Neuroradiol. 36:2394–2399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heggli I, Laux CJ, Mengis T, Karol A,
Cornaz F, Herger N, Aradi-Vegh B, Widmer J, Burkhard MD,
Farshad-Amacker NA, et al: Modic type 2 changes are
fibroinflammatory changes with complement system involvement
adjacent to degenerated vertebral endplates. JOR Spine.
6:e12372022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heggli I, Epprecht S, Juengel A,
Schuepbach R, Farshad-Amacker N, German C, Mengis T, Herger N,
Straumann L, Baumgartner S, et al: Pro-fibrotic phenotype of bone
marrow stromal cells in Modic type 1 changes. Eur Cell Mater.
41:648–667. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu X, Zhu Z, Pan J, Feng Z, Lv X, Battié
MC and Wang Y: Traumatic vertebra and endplate fractures promote
adjacent disc degeneration: Evidence from a clinical MR follow-up
study. Skeletal Radiol. 51:1017–1026. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dudli S, Heggli I, Laux CJ, Spirig JM,
Wanivenhaus F, Betz M, Germann C, Farshad-Amacker NA, Herger N,
Mengis T, et al: Role of C-reactive protein in the bone marrow of
Modic type 1 changes. J Orthop Res. 41:1115–1122. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dudli S, Liebenberg E, Magnitsky S, Miller
S, Demir-Deviren S and Lotz JC: Propionibacterium acnes infected
intervertebral discs cause vertebral bone marrow lesions consistent
with Modic changes. J Orthop Res. 34:1447–1455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biczo A, Szita J, McCall I and Varga PP;
Genodisc Consortium; Lazary A, : Association of vitamin D receptor
gene polymorphisms with disc degeneration. Eur Spine J. 29:596–604.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eksi MS, Kara M, Ozcan-Eksi EE, Aytar MH,
Güngör A, Özgen S and Pamir MN: Is diabetes mellitus a risk factor
for modic changes?: A novel model to understand the association
between intervertebral disc degeneration and end-plate changes. J
Orthop Sci. 25:571–575. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmid G, Witteler A, Willburger R, Kuhnen
C, Jergas M and Koester O: Lumbar disk herniation: Correlation of
histologic findings with marrow signal intensity changes in
vertebral endplates at MR imaging. Radiology. 231:352–358. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roberts S, Evans H, Trivedi J and Menage
J: Histology and pathology of the human intervertebral disc. J Bone
Joint Surg Am. 88 (Suppl 2):S10–S14. 2006. View Article : Google Scholar
|
|
16
|
Feng P, Che Y, Gao C, Zhu L, Gao J and Vo
NV: Immune exposure: How macrophages interact with the nucleus
pulposus. Front Immunol. 14:11557462023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Adams MA, Freeman BJ, Morrison HP, Nelson
IW and Dolan P: Mechanical initiation of intervertebral disc
degeneration. Spine (Phila Pa 1976). 25:1625–1636. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferguson SJ, Ito K and Nolte LP: Fluid
flow and convective transport of solutes within the intervertebral
disc. J Biomech. 37:213–221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Torkki M, Majuri ML, Wolff H, Koskelainen
T, Haapea M, Niinimäki J, Alenius H, Lotz J and Karppinen J:
Osteoclast activators are elevated in intervertebral disks with
Modic changes among patients operated for herniated nucleus
pulposus. Eur Spine J. 25:207–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dudli S, Ferguson SJ and Haschtmann D:
Severity and pattern of post-traumatic intervertebral disc
degeneration depend on the type of injury. Spine J. 14:1256–1264.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dudli S, Haschtmann D and Ferguson SJ:
Fracture of the vertebral endplates, but not equienergetic impact
load, promotes disc degeneration in vitro. J Orthop Res.
30:809–816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jensen TS, Bendix T, Sorensen JS, Manniche
C, Korsholm L and Kjaer P: Characteristics and natural course of
vertebral endplate signal (Modic) changes in the Danish general
population. BMC Musculoskelet Disord. 10:812009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kerttula L, Luoma K, Vehmas T, Gronblad M
and Kaapa E: Modic type I change may predict rapid progressive,
deforming disc degeneration: A prospective 1-year follow-up study.
Eur Spine J. 21:1135–1142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dudli S, Sing DC, Hu SS, Berven SH, Burch
S, Deviren V, Cheng I, Tay BKB, Alamin TF, Ith MAM, et al: Issls
prize in basic science 2017: Intervertebral disc/bone marrow
cross-talk with Modic changes. Eur Spine J. 26:1362–1373. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dudli S, Karol A, Giudici L, Heggli I,
Laux CJ, Spirig JM, Wanivenhaus F, Betz M, Germann C,
Farshad-Amacker N, et al: CD90-positive stromal cells associate
with inflammatory and fibrotic changes in modic changes. Osteoarthr
Cartil Open. 4:1002872022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baum T, Yap SP, Karampinos DC, Nardo L,
Kuo D, Burghardt AJ, Masharani UB, Schwartz AV, Li X and Link TM:
Does vertebral bone marrow fat content correlate with abdominal
adipose tissue, lumbar spine bone mineral density, and blood
biomarkers in women with type 2 diabetes mellitus? J Magn Reson
Imaging. 35:117–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thomas GP, Hemmrich K, Abberton KM,
McCombe D, Penington AJ, Thompson EW and Morrison WA:
Zymosan-induced inflammation stimulates neo-adipogenesis. Int J
Obes (Lond). 32:239–248. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Monden M, Koyama H, Otsuka Y, Morioka T,
Mori K, Shoji T, Mima Y, Motoyama K, Fukumoto S, Shioi A, et al:
Receptor for advanced glycation end products regulates adipocyte
hypertrophy and insulin sensitivity in mice: Involvement of
Toll-like receptor 2. Diabetes. 62:478–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haapasalo K and Meri S: Regulation of the
complement system by pentraxins. Front Immunol. 10:17502019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sjöberg A, Onnerfjord P Mörgelin M,
Heinegård D, Heinegård D and Blom AM: The extracellular matrix and
inflammation: Fibromodulin activates the classical pathway of
complement by directly binding C1q. J Biol Chem. 280:32301–23208.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Webster GF, Leyden JJ and Nilsson UR:
Complement activation in acne vulgaris: Consumption of complement
by comedones. Infect Immun. 26:183–186. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Piccinini AM and Midwood KS: DAMPening
inflammation by modulating TLR signalling. Mediators Inflamm.
2010:6723952010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ignatius A, Schoengraf P, Kreja L, Liedert
A, Recknagel S, Kandert S, Brenner RE, Schneider M, Lambris JD and
Huber-Lang M: Complement C3a and C5a modulate osteoclast formation
and inflammatory response of osteoblasts in synergism with IL-1β. J
Cell Biochem. 112:2594–2605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Llorian-Salvador M, Byrne EM, Szczepan M,
Little K, Chen M and Xu H: Complement activation contributes to
subretinal fibrosis through the induction of
epithelial-to-mesenchymal transition (EMT) in retinal pigment
epithelial cells. J Neuroinflammation. 19:1822022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nozaki M, Raisler BJ, Sakurai E, Sarma JV,
Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ and
Ambati J: Drusen complement components C3a and C5a promote
choroidal neovascularization. Proc Natl Acad Sci USA.
103:2328–2333. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takahashi K, Miyazaki T, Ohnari H, Takino
T and Tomita K: Schmorl's nodes and low-back pain. Analysis of
magnetic resonance imaging findings in symptomatic and asymptomatic
individuals. Eur Spine J. 4:56–59. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dudli S, Fields AJ, Samartzis D, Karppinen
J and Lotz JC: Pathobiology of modic changes. Eur Spine J.
25:3723–3734. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Galán-Díez M and Kousteni S: A bone marrow
niche-derived molecular switch between osteogenesis and
hematopoiesis. Genes Dev. 32:324–326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou BO, Yue R, Murphy MM, Peyer JG and
Morrison SJ: Leptin-receptor-expressing mesenchymal stromal cells
represent the main source of bone formed by adult bone marrow. Cell
Stem Cell. 15:154–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Decker M, Martinez-Morentin L, Wang G, Lee
Y, Liu Q, Leslie J and Ding L: Leptin-receptor-expressing bone
marrow stromal cells are myofibroblasts in primary myelofibrosis.
Nat Cell Biol. 19:677–688. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Papayannopoulos V: Neutrophil
extracellular traps in immunity and disease. Nat Rev Immunol.
18:134–147. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Naveiras O, Nardi V, Wenzel PL, Hauschka
PV, Fahey F and Daley GQ: Bone-marrow adipocytes as negative
regulators of the haematopoietic microenvironment. Nature.
460:259–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wan Y, Chong LW and Evans RM: PPAR-gamma
regulates osteoclastogenesis in mice. Nat Med. 13:1496–1503. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He N, Liu M and Wu Y: Adipose tissue and
hematopoiesis: Friend or foe? J Clin Lab Anal. 37:e248722023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mattiucci D, Maurizi G, Izzi V, Cenci L,
Ciarlantini M, Mancini S, Mensà E, Pascarella R, Vivarelli M,
Olivieri A, et al: Bone marrow adipocytes support hematopoietic
stem cell survival. J Cell Physiol. 233:1500–1511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wilson A, Fu H, Schiffrin M, Winkler C,
Koufany M, Jouzeau JY, Bonnet N, Gilardi F, Renevey F, Luther SA,
et al: Lack of adipocytes alters hematopoiesis in lipodystrophic
mice. Front Immunol. 9:25732018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kolaczkowska E and Kubes P: Neutrophil
recruitment and function in health and inflammation. Nat Rev
Immunol. 13:159–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Borregaard N: Neutrophils, from marrow to
microbes. Immunity. 33:657–670. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Heggli I, Mengis T, Laux CJ, Opitz L,
Herger N, Menghini D, Schuepbach R, Farshad-Amacker NA, Brunner F,
Fields AJ, et al: Low back pain patients with Modic type 1 changes
exhibit distinct bacterial and non-bacterial subtypes. Osteoarthr
Cartil Open. 6:1004342024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kawai T and Akira S: Toll-like receptors
and their crosstalk with other innate receptors in infection and
immunity. Immunity. 34:637–650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fournier BM and Parkos CA: The role of
neutrophils during intestinal inflammation. Mucosal Immunol.
5:354–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thålin C, Hisada Y, Lundström S, Mackman N
and Wallén H: Neutrophil extracellular traps: Villains and targets
in arterial, venous, and cancer-associated thrombosis. Arterioscler
Thromb Vasc Biol. 39:1724–1738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang H, Wang C, Zhao MH and Chen M:
Neutrophil extracellular traps can activate alternative complement
pathways. Clin Exp Immunol. 181:518–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Christoffersson G, Vagesjo E, Vandooren J,
Lidén M, Massena S, Reinert RB, Brissova M, Powers AC, Opdenakker G
and Phillipson M: VEGF-A recruits a proangiogenic MMP-9-delivering
neutrophil subset that induces angiogenesis in transplanted hypoxic
tissue. Blood. 120:4653–4662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shi J, Gilbert GE, Kokubo Y and Ohashi T:
Role of the liver in regulating numbers of circulating neutrophils.
Blood. 98:1226–1230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Okabe Y and Medzhitov R: Tissue biology
perspective on macrophages. Nat Immunol. 17:9–17. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao J, Liang Y and Wang L: Shaping
polarization of tumor-associated macrophages in cancer
immunotherapy. Front Immunol. 13:8887132022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nakazawa KR, Walter BA, Laudier DM,
Krishnamoorthy D, Mosley GE, Spiller KL and Iatridis JC:
Accumulation and localization of macrophage phenotypes with human
intervertebral disc degeneration. Spine J. 18:343–356. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Murai K, Sakai D, Nakamura Y, Nakai T,
Igarashi T, Seo N, Murakami T, Kobayashi E and Mochida J: Primary
immune system responders to nucleus pulposus cells: Evidence for
immune response in disc herniation. Eur Cell Mater. 19:13–21. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Daneshvar A, Nemati P, Azadi A, Amid R and
Kadkhodazadeh M: M2 macrophage-derived exosomes for bone
regeneration: A systematic review. Arch Oral Biol. 166:1060342024.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Fan S, Zhang C, Sun X, Su C, Xue Y, Song X
and Deng R: Metformin enhances osteogenic differentiation of BMSC
by modulating macrophage M2 polarization. Sci Rep. 14:202672024.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fang C, Zhong R, Lu S, Yu G, Liu Z, Yan C,
Gao J, Tang Y, Wang Y, Zhao Q and Feng X: TREM2 promotes macrophage
polarization from M1 to M2 and suppresses osteoarthritis through
the NF-κB/CXCL3 axis. Int J Biol Sci. 20:1992–2007. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang Y, Zhao M, Liu S, Guo J, Lu Y, Cheng
J and Liu J: Macrophage-derived extracellular vesicles: Diverse
mediators of pathology and therapeutics in multiple diseases. Cell
Death Dis. 11:9242020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Häusler KD, Horwood NJ, Chuman Y, Fisher
JL, Ellis J, Martin TJ, Rubin JS and Gillespie MT: Secreted
frizzled-related protein-1 inhibits RANKL-dependent osteoclast
formation. J Bone Miner Res. 19:1873–1881. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang Y, Qin J, Lan L, Li N, Wang C, He P,
Liu F, Ni H and Wang Y: M-CSF cooperating with NFκB induces
macrophage transformation from M1 to M2 by upregulating c-Jun.
Cancer Biol Ther. 15:99–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ikebuchi Y, Aoki S, Honma M, Hayashi M,
Sugamori Y, Khan M, Kariya Y, Kato G, Tabata Y, Penninger JM, et
al: Coupling of bone resorption and formation by RANKL reverse
signalling. Nature. 561:195–200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pajarinen J, Lin T, Gibon E, Kohno Y,
Maruyama M, Nathan K, Lu L, Yao Z and Goodman SB: Mesenchymal stem
cell-macrophage crosstalk and bone healing. Biomaterials.
196:80–89. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ni S, Ling Z, Wang X, Cao Y, Wu T, Deng R,
Crane JL, Skolasky R, Demehri S, Zhen G, et al: Sensory innervation
in porous endplates by Netrin-1 from osteoclasts mediates
PGE2-induced spinal hypersensitivity in mice. Nat Commun.
10:56432019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bachmeier BE, Nerlich AG, Weiler C,
Paesold G, Jochum M and Boos N: Analysis of tissue distribution of
TNF-alpha, TNF-alpha-receptors, and the activating
TNF-alpha-converting enzyme suggests activation of the TNF-alpha
system in the aging intervertebral disc. Ann N Y Acad Sci.
1096:44–54. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wallach D: The TNF family: Only the
surface has been scratched. Semin Immunol. 26:181–182. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Aggarwal BB, Gupta SC and Kim JH:
Historical perspectives on tumor necrosis factor and its
superfamily: 25 years later, a golden journey. Blood. 119:651–665.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang J, Tian Y, Phillips KL, Chiverton N,
Haddock G, Bunning RA, Cross AK, Shapiro IM, Le Maitre CL and
Risbud MV: Tumor necrosis factor alpha- and
interleukin-1beta-dependent induction of CCL3 expression by nucleus
pulposus cells promotes macrophage migration through CCR1.
Arthritis Rheum. 65:832–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Nakawaki M, Uchida K, Miyagi M, Inoue G,
Kawakubo A, Kuroda A, Satoh M and Takaso M: Sequential CCL2
expression profile after disc injury in mice. J Orthop Res.
38:895–901. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chou PH, Chee A, Shi P, Lin CL, Zhao Y,
Zhang L and An HS: Small molecule antagonist of C-C chemokine
receptor 1 (CCR1) reduces disc inflammation in the rabbit model.
Spine J. 20:2025–2036. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Li Z, Wang X, Pan H, Yang H, Li X, Zhang
K, Wang H, Zheng Z, Liu H and Wang J: Resistin promotes CCL4
expression through toll-like receptor-4 and activation of the
p38-MAPK and NF-ĸB signaling pathways: Implications for
intervertebral disc degeneration. Osteoarthritis Cartilage.
25:341–350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hwang MH, Son HG, Lee JW, Yoo CM, Shin JH,
Nam HG, Lim HJ, Baek SM, Park JH, Kim JH and Choi H:
Photobiomodulation of extracellular matrix enzymes in human nucleus
pulposus cells as a potential treatment for intervertebral disk
degeneration. Sci Rep. 8:116542018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Abe Y, Akeda K, An HS, Aoki Y, Pichika R,
Muehleman C, Kimura T and Masuda K: Proinflammatory cytokines
stimulate the expression of nerve growth factor by human
intervertebral disc cells. Spine (Phila Pa 1976). 32:635–642. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dudli S, Liebenberg E, Magnitsky S, Lu B,
Lauricella M and Lotz JC: Modic type 1 change is an autoimmune
response that requires a proinflammatory milieu provided by the
‘Modic disc’. Spine J. 18:831–844. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jia J, Nie L and Liu Y: Butyrate
alleviates inflammatory response and NF-ĸB activation in human
degenerated intervertebral disc tissues. Int Immunopharmacol.
78:1060042020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ma Z, Tang P, Dong W, Lu Y, Tan B, Zhou N,
Hao J, Shen J and Hu Z: SIRT1 alleviates IL-1β induced nucleus
pulposus cells pyroptosis via mitophagy in intervertebral disc
degeneration. Int Immunopharmacol. 107:1086712022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang K, Ding W, Sun W, Sun XJ, Xie YZ,
Zhao CQ and Zhao J: Beta1 integrin inhibits apoptosis induced by
cyclic stretch in annulus fibrosus cells via ERK1/2 MAPK pathway.
Apoptosis. 21:13–24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kang H, Dong Y, Peng R, Liu H, Guo Q, Song
K, Zhu M, Yu K, Wu W and Li F: Inhibition of IRE1 suppresses the
catabolic effect of IL-1β on nucleus pulposus cell and prevents
intervertebral disc degeneration in vivo. Biochem Pharmacol.
197:1149322022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhao F, Guo Z, Hou F, Fan W, Wu B and Qian
Z: Magnoflorine alleviates ‘M1’ polarized macrophage-induced
intervertebral disc degeneration through repressing the
HMGB1/Myd88/NF-ĸB pathway and NLRP3 inflammasome. Front Pharmacol.
12:7010872021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang S, Wang P, Hu B, Liu W, Lv X, Chen S
and Shao Z: HSP90 inhibitor 17-AAG attenuates nucleus pulposus
inflammation and catabolism induced by M1-polarized macrophages.
Front Cell Dev Biol. 9:7969742022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang S, Wang P, Hu B, Lv X, Liu W, Chen S
and Shao Z: Inhibiting heat shock protein 90 attenuates nucleus
pulposus fibrosis and pathologic angiogenesis induced by
macrophages via down-regulating cell migration-inducing protein. Am
J Pathol. 193:960–976. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
England H, Summersgill HR, Edye ME,
Rothwell NJ and Brough D: Release of interleukin-1alpha or
interleukin-1beta depends on mechanism of cell death. J Biol Chem.
289:15942–15950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Phillips KL, Jordan-Mahy N, Nicklin MJ and
Le Maitre CL: Interleukin-1 receptor antagonist deficient mice
provide insights into pathogenesis of human intervertebral disc
degeneration. Ann Rheum Dis. 72:1860–1867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hasvik E, Schjolberg T, Jacobsen DP,
Haugen AJ, Grøvle L, Schistad EI and Gjerstad J: Up-regulation of
circulating microRNA-17 is associated with lumbar radicular pain
following disc herniation. Arthritis Res Ther. 21:1862019.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cui S and Zhang L: microRNA-129-5p
shuttled by mesenchymal stem cell-derived extracellular vesicles
alleviates intervertebral disc degeneration via blockade of
LRG1-mediated p38 MAPK activation. J Tissue Eng.
12:204173142110216792021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhao X, Sun Z, Xu B, Duan W, Chang L, Lai
K and Ye Z: Degenerated nucleus pulposus cells derived exosome
carrying miR-27a-3p aggravates intervertebral disc degeneration by
inducing M1 polarization of macrophages. J Nanobiotechnology.
21:3172023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Murray PJ: Macrophage polarization. Annu
Rev Physiol. 79:541–566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yamamoto Y, Kokubo Y, Nakajima H, Honjoh
K, Watanabe S and Matsumine A: Distribution and polarization of
hematogenous macrophages associated with the progression of
intervertebral disc degeneration. Spine (Phila Pa 1976).
47:E149–E158. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Greenlee-Wacker MC: Clearance of apoptotic
neutrophils and resolution of inflammation. Immunol Rev.
273:357–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ip WKE, Hoshi N, Shouval DS, Snapper S and
Medzhitov R: Anti-inflammatory effect of IL-10 mediated by
metabolic reprogramming of macrophages. Science. 356:513–519. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Odegaard JI, Ricardo-Gonzalez RR, Goforth
MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D,
Brombacher F, Ferrante AW and Chawla A: Macrophage-specific
PPARgamma controls alternative activation and improves insulin
resistance. Nature. 447:1116–1120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Szanto A, Balint BL, Nagy ZS, Barta E,
Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, et al: STAT6
transcription factor is a facilitator of the nuclear receptor
PPARγ-Regulated gene expression in macrophages and dendritic cells.
Immunity. 33:699–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kawakubo A, Miyagi M, Yokozeki Y, Nakawaki
M, Takano S, Satoh M, Itakura M, Inoue G, Takaso M and Uchida K:
Origin of M2 Mϕ and its macrophage polarization by TGF-β in a mice
intervertebral injury model. Int J Immunopathol Pharmacol.
36:39463202211037922022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xu YQ, Zhang ZH, Zheng YF and Feng SQ:
Dysregulated miR-133a mediates loss of type II collagen by directly
targeting matrix metalloproteinase 9 (MMP9) in human intervertebral
disc degeneration. Spine (Phila Pa 1976). 41:E717–E724. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kobayashi T, Kim H, Liu X, Sugiura H,
Kohyama T, Fang Q, Wen FQ, Abe S, Wang X, Atkinson JJ, et al:
Matrix metalloproteinase-9 activates TGF-beta and stimulates
fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol
Physiol. 306:L1006–L1015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
van Caam A, Vonk M, van den Hoogen F, van
Lent P and van der Kraan P: Unraveling SSc Pathophysiology; The
Myofibroblast. Front Immunol. 9:24522018. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Abbott RD, Purmessur D, Monsey RD,
Brigstock DR, Laudier DM and Iatridis JC: Degenerative grade
affects the responses of human nucleus pulposus cells to link-N,
CTGF, and TGFβ3. J Spinal Disord Tech. 26:E86–E94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wei Q, Liu D, Chu G, Yu Q, Liu Z, Li J,
Meng Q, Wang W, Han F and Li B: TGF-β1-supplemented decellularized
annulus fibrosus matrix hydrogels promote annulus fibrosus repair.
Bioact Mater. 19:581–593. 2022.PubMed/NCBI
|
|
103
|
Zhu L, Yang Y, Yan Z, Zeng J, Weng F, Shi
Y, Shen P, Liu L and Yang H: Controlled release of TGF-β3 for
effective local endogenous repair in IDD using rat model. Int J
Nanomedicine. 17:2079–2096. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Montgomery SR, Nargizyan T, Meliton V,
Nachtergaele S, Rohatgi R, Stappenbeck F, Jung ME, Johnson JS,
Aghdasi B, Tian H, et al: A novel osteogenic oxysterol compound for
therapeutic development to promote bone growth: activation of
hedgehog signaling and osteogenesis through smoothened binding. J
Bone Miner Res. 29:1872–1885. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jacobsen CM, Schwartz MA, Roberts HJ, Lim
KE, Spevak L, Boskey AL, Zurakowski D, Robling AG and Warman ML:
Enhanced Wnt signaling improves bone mass and strength, but not
brittleness, in the Col1a1(+/mov13) mouse model of type I
Osteogenesis Imperfecta. Bone. 90:127–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ramasamy SK, Kusumbe AP, Wang L and Adams
RH: Endothelial notch activity promotes angiogenesis and
osteogenesis in bone. Nature. 507:376–380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Heggli I, Teixeira GQ, Iatridis JC,
Neidlinger-Wilke C and Dudli S: The role of the complement system
in disc degeneration and Modic changes. JOR Spine. 7:e13122024.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Komori T: Cell death in chondrocytes,
osteoblasts, and osteocytes. Int J Mol Sci. 17:20452016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Albert HB, Lambert P, Rollason J, Sorensen
JS, Worthington T, Pedersen MB, Nørgaard HS, Vernallis A, Busch F,
Manniche C and Elliott T: Does nuclear tissue infected with
bacteria following disc herniations lead to Modic changes in the
adjacent vertebrae? Eur Spine J. 22:690–696. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Riegger J, Huber-Lang M and Brenner RE:
Crucial role of the terminal complement complex in chondrocyte
death and hypertrophy after cartilage trauma. Osteoarthritis
Cartilage. 28:685–697. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fatoba O, Itokazu T and Yamashita T:
Complement cascade functions during brain development and
neurodegeneration. FEBS J. 289:2085–2109. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Koivisto K, Jarvinen J, Karppinen J,
Haapea M, Paananen M, Kyllönen E, Tervonen O and Niinimäki J: The
effect of zoledronic acid on type and volume of Modic changes among
patients with low back pain. BMC Musculoskelet Disord. 18:2742017.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xu L, Chu B, Feng Y, Xu F and Zou YF:
Modic changes in lumbar spine: Prevalence and distribution patterns
of end plate oedema and end plate sclerosis. Br J Radiol.
89:201506502016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Galbusera F, Casaroli G and Bassani T:
Artificial intelligence and machine learning in spine research. JOR
Spine. 2:e10442019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gao KT, Tibrewala R, Hess M, Bharadwaj UU,
Inamdar G, Link TM, Chin CT, Pedoia V and Majumdar S: Automatic
detection and voxel-wise mapping of lumbar spine Modic changes with
deep learning. JOR Spine. 5:e12042022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Rajasekaran S, Bt P, Murugan C, Mengesha
MG, Easwaran M, Naik AS, Ks SVA, Kanna RM and Shetty AP: The
disc-endplate-bone-marrow complex classification: progress in our
understanding of Modic vertebral endplate changes and their
clinical relevance. Spine J. 24:34–45. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Laustsen AF and Bech-Azeddine R: Do Modic
changes have an impact on clinical outcome in lumbar spine surgery?
A systematic literature review. Eur Spine J. 25:3735–3745. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Määttä JH, Wadge S, MacGregor A, Karppinen
J and Williams FM: ISSLS prize winner: Vertebral endplate (Modic)
change is an independent risk factor for episodes of severe and
disabling low back pain. Spine (Phila Pa 1976). 40:1187–1193. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jensen TS, Karppinen J, Sorensen JS,
Niinimäki J and Leboeuf-Yde C: Vertebral endplate signal changes
(Modic change): A systematic literature review of prevalence and
association with non-specific low back pain. Eur Spine J.
17:1407–1422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Lambrechts MJ, Brush P, Issa TZ, Toci GR,
Heard JC, Syal A, Schilken MM, Canseco JA, Kepler CK and Vaccaro
AR: Evaluating the impact of modic changes on operative treatment
in the cervical and Lumbar Spine: A systematic review and
meta-analysis. Int J Environ Res Public Health. 19:101582022.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Nian S, Li N, Kong F, Lu S and Chen J: Is
discectomy effective for treating low back pain in patients with
lumbar disc herniation and Modic changes? A systematic review and
meta-analysis of cohort studies. Spine J. 23:533–549. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Conger A, Burnham TR, Clark T, Teramoto M
and McCormick ZL: The effectiveness of intraosseous basivertebral
nerve radiofrequency ablation for the treatment of vertebrogenic
low back pain: An updated systematic review with single-arm
meta-analysis. Pain Med. 23 (Suppl 2):S50–S62. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ohtori S, Inoue G, Ito T, Koshi T, Ozawa
T, Doya H, Saito T, Moriya H and Takahashi K: Tumor necrosis
factor-immunoreactive cells and PGP 9.5-immunoreactive nerve fibers
in vertebral endplates of patients with discogenic low back pain
and modic type 1 or type 2 changes on MRI. Spine (Phila Pa 1976).
31:1026–1031. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
DePalma MJ, Ketchum JM and Saullo T: What
is the source of chronic low back pain and does age play a role?
Pain Med. 12:224–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Depalma MJ, Ketchum JM, Trussell BS,
Saullo TR and Slipman CW: Does the location of low back pain
predict its source? PM R. 3:33–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Feng Z, Liu Y, Yang G, Battie MC and Wang
Y: Lumbar vertebral endplate defects on magnetic resonance images:
Classification, distribution patterns, and associations with modic
changes and disc degeneration. Spine (Phila Pa 1976). 43:919–927.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Udby PM, Samartzis D, Carreon LY, Andersen
MØ, Karppinen J and Modic M: A definition and clinical grading of
Modic changes. J Orthop Res. 40:301–307. 2022. View Article : Google Scholar : PubMed/NCBI
|