Introduction
Cytoplasmic phospholipase A2 (cPLA2; Fig. 1) belongs to the lipolytic enzyme
family and hydrolyzes the ester bond at the sn-2 position of
phospholipids, generating free fatty acids and lysophospholipids.
Members of the PLA2 family are classified into six subfamilies
based on their location in the body, substrate specificity and
physiological functions: Secretory PLA2s (sPLA2s), cPLA2s,
Ca2+-independent PLA2s (iPLA2s), platelet-activating
factor acetylhydrolase PLA2s (PAF-AH PLA2s), lysosomal PLA2s
(LPLA2s) and adipose tissue-specific PLA2s (AdPLA2s). Among these,
cPLA2s play a crucial role in inflammatory diseases, cerebral
ischemia-reperfusion injury, hypertension and autoimmune diseases
(1–4).
cPLA2, a member of the PLA2 family, is a
single-subunit protein composed of 749 amino acids with two
functional domains: the Ca2+-dependent lipid-binding
(CaLB) domain, also known as the C2 domain and the catalytic active
region (CAT), connected by flexible hinges. cPLA2 primarily acts on
the phospholipid bilayer of cell membranes to catalyze the release
of free arachidonic acid (AA) (5).
cPLA2 is activated through two primary pathways: The
Ca2+-dependent pathway, when intracellular
Ca2+ levels rise, Ca2+ binds to the C2 domain
of cPLA2, facilitating its translocation from the cytoplasm to the
membrane's phospholipid matrix; the phosphorylation pathway, when
phosphorylation of amino acid residues in the hinge region between
the C2 domain and the catalytic domain enhances the binding
affinity and catalytic efficiency of cPLA2. This phosphorylation
induces conformational changes, bringing the catalytic domain
closer to the substrate. Ultimately, cPLA2 on the phospholipid
membrane catalyzes phosphatidylinositol hydrolysis, promoting AA
release (6,7). AA, an essential fatty acid, can be
metabolized into various bioactive substances, including
prostaglandins, platelet-activating factors and leukotrienes, which
regulate pathophysiological processes such as inflammation, cell
proliferation, apoptosis and platelet aggregation (8,9).
cPLA2 has been shown to participate in essential cellular
processes, including phospholipid metabolism, signal transduction
and membrane remodeling under physiological conditions (1,10,11).
However, increased cPLA2 activity and excessive AA release, along
with pro-inflammatory mediators, can compromise lysosomal membrane
integrity, exacerbating inflammation and oxidative stress under
pathological conditions (12).
Numerous studies have confirmed the involvement of cPLA2 in the
pathogenesis of various diseases. However, systematic summaries of
its role in cardiovascular diseases and underlying mechanisms
remain limited. Existing evidence indicates that cPLA2 plays a
pivotal role in the development of cardiovascular conditions,
including atherosclerosis, myocardial ischemia-reperfusion injury
and hypertension (3,4,13)
(Table I). This review highlighted
the functional significance and potential mechanisms of cPLA2 in
cardiovascular diseases, offering novel insights into their
diagnosis and treatment.
 | Table I.Roles and mechanisms of cPLA2 in
various cardiovascular diseases. |
Table I.
Roles and mechanisms of cPLA2 in
various cardiovascular diseases.
| Cardiovascular
diseases | cPLA2-related
pathogenic mechanisms | (Refs.) |
|---|
|
Atherosclerosis | Inflammatory
responses and oxidative stress lead to increased vascular
permeability, while the proliferation and migration of smooth
muscle cells contribute to plaque formation and vascular
remodeling. Excessive cell apoptosis results in endothelial cell
damage, accompanied by a blockade of autophagy flux, which causes
the accumulation of harmful substances and damaged organelles. | (13,28,29,32–35,56–60) |
| Coronary artery
disease | Inflammatory
responses provoke plaque instability and rupture, coupled with
platelet activation that causes platelet aggregation and thrombus
formation. | (28,60) |
| Myocardial
infarction | Inflammatory
responses and oxidative stress inflict damage on myocardial cells.
This is exacerbated by excessive cell apoptosis during reperfusion
injury and platelet activation, which together promote plaque and
thrombus formation. The process is further complicated by a
blockade of autophagy flux, leading to the accumulation of harmful
substances and damaged organelles. | (28,29,32–35,56–60) |
| Heart failure | The damage to
myocardial cells from inflammatory responses and oxidative stress
necessitates myocardial remodeling, involving cell apoptosis,
myocardial fibrosis and hypertrophy. This scenario is compounded by
the blockade of autophagy flux, resulting in the accumulation of
harmful substances and damaged organelles. | (32–35,56–60) |
| Hypertension | Inflammatory
responses and oxidative stress cause endothelial dysfunction
through inflammatory mediators, with subsequent vasoconstriction
regulated by sodium and calcium channels. This leads to
arteriosclerosis, which narrows blood vessels and is regulated
further by the renin-angiotensin system. | (13,67,74) |
| Cardiac valve
disease | Inflammatory
responses and oxidative stress lead to the destruction and
proliferation of valve tissue. This promotes valve cell
proliferation and migration, resulting in the thickening,
calcification and fibrosis of the valves. | (1,32–34,75) |
cPLA2 exacerbates inflammation
cPLA2 promotes the production and
release of inflammatory mediators
Tissue damage and oxidative stress can activate
cPLA2, which hydrolyzes the sn-2 site of membrane phospholipids to
produce metabolites, primarily AA and lysophospholipids. These
metabolites, in turn, generate downstream molecules such as
leukotrienes, prostaglandins, lipoxin A, thromboxane, sphingomyelin
and lysophosphatidic acid via cyclooxygenase (COX) and
lysophospholipase activity. These molecules trigger inflammatory
responses and oxidative stress, resulting in a robust inflammatory
cascade (14).
cPLA2 activates the inflammatory
signal conduction pathway
Key signaling pathways such as NF-κB and MAPKs play
critical roles in cell signal transmission, regulating processes
such as inflammation, immune responses, cell proliferation and
apoptosis. Studies have confirmed that cPLA2 activates these
inflammatory signaling pathways (1,15–18)
(Fig. 2).
NF-κB signaling pathway
NF-κB is a classical transcription factor that
regulates the expression of multiple genes by translocating from
the cytoplasm to the nucleus. It serves as a key regulator of
inflammatory responses, influencing both the progression and
resolution of inflammation. AA released by activated cPLA2 acts as
a second messenger, participating in downstream signaling
activation. AA is catalyzed by COX into prostaglandin H2 (PGH2),
which is subsequently converted into prostaglandin E2 (PGE2). PGE2
activates G protein-coupled receptors (GPCRs) on cell membranes,
triggering downstream signaling events such as the phosphorylation
and degradation of IκB (inhibitor of NF-κB). The degradation of IκB
releases active NF-κB, promoting its nuclear translocation and
binding to specific DNA sequences to initiate the transcription of
downstream genes. Excessive activation of cPLA2 can prolong NF-κB
activity, leading to sustained inflammatory responses and tissue
damage (15–17).
MAPKs signaling pathway
Mitogen-activated protein kinases (MAPKs) represent
a highly conserved cell signal transduction pathway that conveys
extracellular and intracellular signals to regulatory networks
through phosphorylation of key protein targets. The primary
components include ERK1/2, JNK and p38 MAPK (18). The MAPKs pathway can be activated
by various exogenous stimuli, such as growth factors, stress and
inflammatory factors, or endogenous stimuli, such as cellular
stress and DNA damage. Stimulation leads to the activation of
receptors, such as receptor tyrosine kinases and GPCRs, which
subsequently activate MAP3K through a series of downstream kinase
cascades. MAP3K phosphorylates MAP2K, which, in turn, activates
MAPK. This process triggers the activation of nuclear transcription
factors, ultimately regulating cellular physiological functions
(19). cPLA2 activates the MAPKs
signaling pathway through several mechanisms. Diacylglycerol,
generated by cPLA2-catalyzed phosphatidylinositol 2 (PIP2),
directly activates protein kinase C, which then stimulates the
MAPKs pathway (20). Additionally,
AA, a product of cPLA2 catalysis, plays a crucial role in the MAPKs
network. AA activates ERK, JNK and p38 MAPK, influencing processes
such as cell proliferation, apoptosis and differentiation. Among
these, p38 MAPK primarily mediates inflammatory responses and
cellular stress (18,21). In summary, cPLA2 serves as a key
upstream molecule in the activation of MAPKs signaling
pathways.
The PI3K/Akt signaling pathway
cPLA2, as a phosphatidylinositol-specific
phosphoesterase, activates the PI3K/Akt signaling pathway through
multiple mechanisms. It promotes this pathway by inducing the
production and release of growth factors. For instance, cPLA2
facilitates the secretion of TNF-α, which activates the PI3K/Akt
pathway (21). Moreover, cPLA2
enhances PI3K activity by regulating phosphatidylinositol content
in cell membranes, thereby increasing the affinity of PI3K
(22–24). Additionally, cPLA2 directly
activates Akt by promoting its phosphorylation and enhancing its
activity, which further drives the PI3K/Akt signaling pathway
(25).
cPLA2 participates in platelet
activation
A number of studies have demonstrated that cPLA2
plays a pivotal role in platelet activation. Platelet activation,
typically induced by external stimuli such as cytokines or vascular
damage, triggers intracellular signaling pathways that activate
cPLA2, leading to the release of AA. In platelets, AA is primarily
metabolized by COX-1 into PGH2, which is subsequently converted
into thromboxane A2 (TXA2) (26).
TXA2 promotes platelet activation and vasoconstriction (27). Upon activation, platelets release
bioactive substances such as platelet factor 4, platelet-derived
growth factor and ADP. These substances stimulate neighboring
platelets, enhancing adhesion and aggregation. Additionally,
activated platelets expose receptors, including GPIIb/IIIa and
GPIb/IX, on the endothelial surface. These receptors bind to
molecules such as fibronectin and von Willebrand factors, exposed
during vascular injury, further facilitating platelet aggregation
and the formation of platelet thrombi (28,29).
PI3K/Akt and MAPK pathways are the two major signaling pathways
involved in platelet activation and aggregation. The PI3K/Akt
pathway is activated by various platelet agonists, including
thrombin, collagen and ADP, promoting platelet activation, granule
release and integrin activation. Similarly, the MAPK pathway is
activated by platelet agonists and regulates genes involved in
platelet functions, such as integrin and TXA production, thereby
enhancing platelet activation and aggregation (30,31).
In summary, cPLA2 is a critical regulator of platelet activation,
functioning through its involvement in the PI3K/Akt and MAPK
signaling pathways.
cPLA2 regulates myocardial cell
apoptosis
In the study of cell death, several mechanisms have
been recognized, including apoptosis, necroptosis, pyroptosis and
ferroptosis. These mechanisms play critical roles in both
physiological and pathological cellular processes (32). cPLA2 plays a pivotal regulatory
role in cell death by catalyzing the hydrolysis of cell membrane
phospholipids to generate arachidonic acid and its metabolic
products (33). In necroptosis,
cPLA2 facilitates membrane remodeling and the release of necrotic
signals by regulating lipid metabolism and altering cell membrane
structure (34). In pyroptosis,
cPLA2 activates the synthesis of pro-inflammatory cytokines, such
as IL-1β and IL-18, by releasing AA, which further promotes
inflammasome formation and triggers the pyroptotic response
(35). Additionally, during
ferroptosis, cPLA2 modulates membrane lipid peroxidation,
influencing oxidative damage to membrane lipids and the metabolism
of intracellular iron, thereby indirectly regulating ferroptosis
(36). The present review focused
on the critical role of cPLA2 in apoptosis, specifically its
regulation of the multiple signaling pathways involved in this
process.
Apoptosis is the most well-studied and classical
form of programmed cell death, crucial for maintaining normal
tissue structure and function. Excessive apoptosis of myocardial
cells results in significant cell loss, leading to myocardial
tissue depletion and impaired blood supply to the affected area
(37,38). This process triggers cardiac
fibrosis, where healthy myocardium is replaced by fibrous
connective tissue. Fibrosis renders the heart stiff, reduces its
elasticity and causes ventricular dilation. These changes
exacerbate cardiac remodeling, further impairing cardiac function
and leading to alterations in the overall structure of the heart
(39). Excessive apoptosis has
been implicated in myocardial ischemia, ischemia/reperfusion
injury, post-ischemic cardiac remodeling and the progression of
cardiovascular conditions such as coronary atherosclerosis,
myocardial infarction, hypertension and heart failure (40–44).
The following paragraphs elaborate on these processes (Fig. 2).
MAPKs signaling
cPLA2 and MAPK signaling pathways are closely linked
in the regulation of cell apoptosis. cPLA2 participates in the
metabolism of PIP2 on the cell membrane, converting it into
phosphatidylinositol triphosphate (PIP3) (2). PIP3 plays a crucial role in the
apoptotic process. cPLA2 activates PI3K through PIP3, which
subsequently activates MAPKKK, further stimulating the MAPK
signaling pathway (41). The MAPK
signaling pathway promotes apoptosis in various cell types,
including cardiomyocytes, endothelial cells, macrophages and tumor
cells, through different subtypes (such as JNK, p38 MAPK and ERK).
Activated JNK and p38 MAPK enhance the expression of
apoptosis-related transcription factors (such as p53) and increase
pro-apoptotic genes (such as Bax), while simultaneously
downregulating anti-apoptotic factors such as Bcl-2) (42,45).
In cardiomyocyte apoptosis, distinct MAPK members may have varying
roles (46). JNK activation
regulates cell apoptosis by either stimulating apoptotic factor
expression or inhibiting anti-apoptotic mechanisms. For instance,
JNK activates the transcription factor c-Jun, promoting the
expression of apoptosis-related genes, including apoptotic proteins
and mitochondrial regulatory factors, ultimately leading to cell
apoptosis in cardiomyocytes (47).
However, JNK activation may act as an anti-apoptotic factor in
cardiomyocytes derived from embryonic stem cells (48). Under pathological conditions such
as ischemia-reperfusion, inflammation and oxidative stress, p38
activation increases mitochondrial membrane permeability and
caspase enzyme activation, thereby promoting myocardial cell
apoptosis (49). ERK, through a
series of cascade reactions, plays a protective role in myocardial
cell apoptosis. ERK activation inhibits cardiomyocyte apoptosis by
upregulating the Bcl-2/Bax ratio through downregulation of Bax
expression, thereby maintaining mitochondrial stability (50). In summary, cPLA2 regulates
myocardial cell apoptosis by activating different members of the
MAPK signaling pathway.
PI3K/Akt signaling
The PI3K/Akt signaling pathway plays a crucial role
in regulating cell survival and apoptosis, primarily through its
control of anti-apoptotic mechanisms. Dysregulation of this pathway
is closely associated with the onset and progression of various
diseases, particularly cancer, neurodegenerative disorders and
cardiovascular diseases (51,52).
Akt is the primary effector kinase, regulating cell survival and
apoptosis by phosphorylating a series of downstream target
proteins. Akt reduces the pro-apoptotic effects of factors such as
Bad and Bax by phosphorylating and inhibiting them. It also
phosphorylates and activates anti-apoptotic proteins, such as Bcl-2
and Bcl-xL, thereby enhancing cell survival (53). The PI3K/Akt signaling pathway is
essential for the normal physiological functions of cardiomyocytes
and is closely related to cardiomyocyte apoptosis. Dysregulation of
this pathway can lead to cardiomyocyte apoptosis, contributing to
cardiovascular diseases, including myocardial infarction and heart
failure (52). Activation of the
PI3K/Akt pathway promotes myocardial cell survival and inhibits
apoptosis (49). The PI3K/Akt
pathway regulates the release of AA and associated apoptosis
processes by inhibiting cPLA2 activity. Activated Akt directly
phosphorylates and inhibits cPLA2, reducing AA production (43). Additionally, Akt can regulate cPLA2
activity by preventing its translocation to the cell membrane, a
critical step in AA release. Akt signaling disrupts this process,
thereby decreasing AA production. However, under certain
pathological conditions, such as myocardial ischemia, injury, or
hypertrophy, the PI3K/Akt signaling pathway is inhibited. This
inhibition weakens the effect of Akt on cPLA2, potentially leading
to cPLA2 activation, increased AA release and enhanced cell
apoptosis (54).
NF-κB signaling
As aforementioned, cPLA2 activates the NF-κB
signaling pathway by producing AA and its metabolites. This pathway
promotes cell apoptosis by regulating the expression of apoptotic
factors. NF-κB induces FasL expression, initiating apoptotic
signaling and promoting cell apoptosis. It may also increase the
production of TNF-α, which activates downstream apoptotic signals,
including Caspase-8, to initiate apoptosis (44). Additionally, NF-κB accelerates cell
apoptosis by regulating p53, further exacerbating cardiac damage
(55). These mechanisms play a
significant role in various cardiovascular diseases, including
heart failure and coronary artery disease (56). For example, NF-κB mediates
atherosclerosis through several key mechanisms: First, it promotes
endothelial cells to express pro-inflammatory molecules, recruiting
and activating inflammatory cells to enhance local inflammation.
Second, NF-κB affects the proliferation and migration of smooth
muscle cells, contributing to plaque formation. It also increases
oxidative stress, leads to endothelial injury and promotes the
formation of foam cells through the accumulation of cholesterol and
lipids. These processes collectively promote the development of
atherosclerosis (13,56).
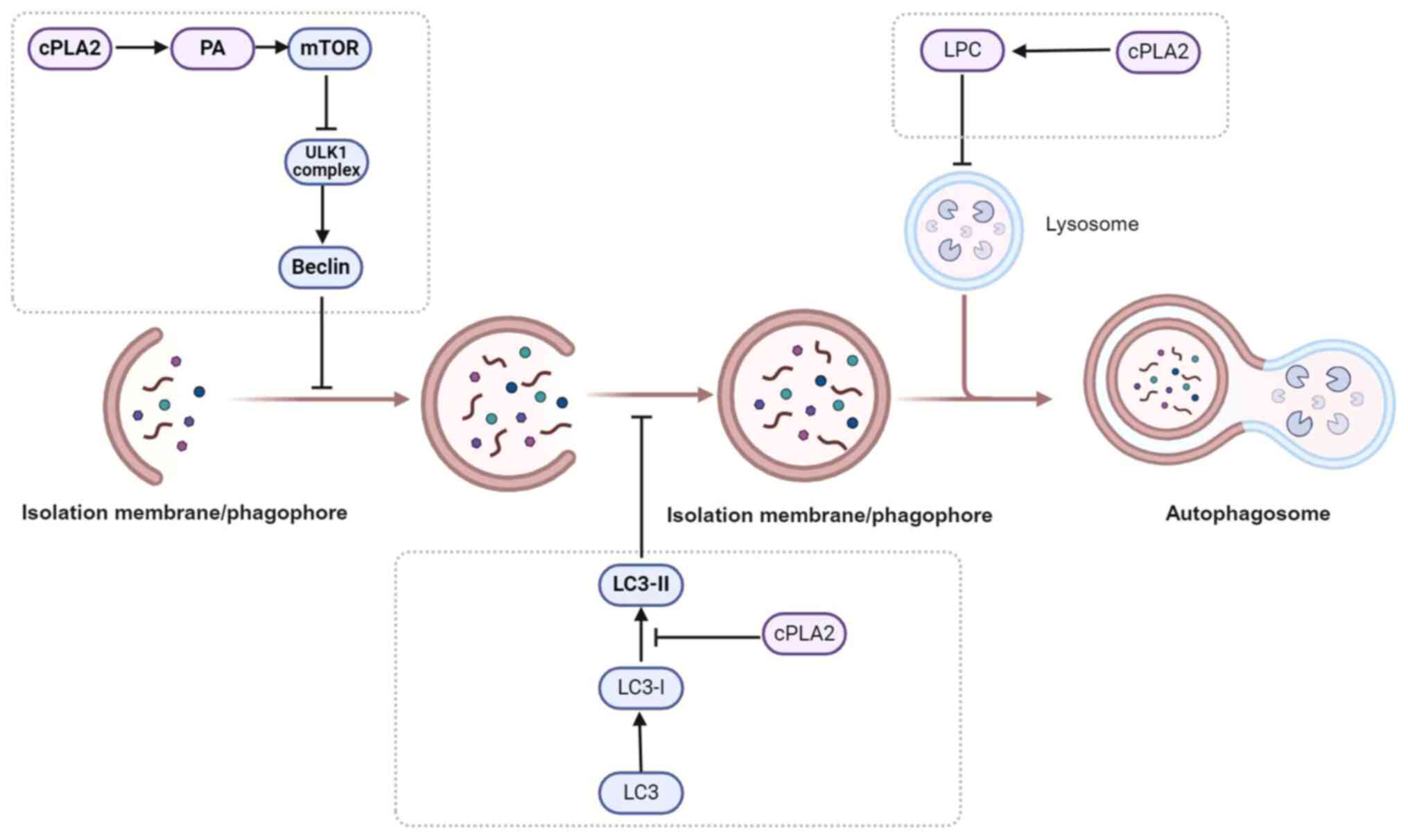
cPLA2 participates in the autophagy
flux
Autophagy is a process of cellular self-degradation
that maintains cellular homeostasis by degrading and recycling
harmful or aging components, allowing cells to adapt to
environmental changes (57). This
process involves three main stages: Activation, transportation and
degradation. Specifically, it includes the formation of
phagophores, the development of autophagosomes, the fusion of
autophagosomes and lysosomes to form autolysosomes and the
subsequent degradation of substrates within them (58). This entire process is referred to
as autophagic flux. Studies have shown that autophagy plays a
crucial role in various cardiovascular diseases. The heart is a
highly metabolically active organ with significant demands for
oxygen and energy. Under conditions such as ischemia, reperfusion
injury, or other pathological states, autophagy can be activated to
meet energy needs, reduce oxidative stress, inhibit cell apoptosis
and maintain cellular homeostasis, thereby protecting the heart
from damage (59,60). In heart diseases such as myocardial
infarction and heart failure, autophagic flux is often inhibited or
impaired. This abnormal autophagic activity leads to the
accumulation of harmful substances and damaged organelles within
myocardial cells, further exacerbating cell damage (61,62).
Additionally, studies using acute hemodynamic stress models have
shown that excessive autophagy can result in increased myocardial
cell hypertrophy, impaired cardiac performance and the activation
of cardiac stromal cells (such as fibroblasts), promoting fibrosis.
These mechanisms suggest that excessive autophagy can have
pathological consequences, potentially contributing to cardiac
hypertrophy and heart failure (63,64).
Furthermore, autophagic flux dysfunction can reduce endothelial
cell tolerance to oxidative stress and inflammatory responses,
leading to the accumulation of oxidized LDL (low-density
lipoprotein) and promoting the formation and progression of
atherosclerosis. Excessive autophagy may also increase cell
apoptosis within plaques, compromising their structural stability
and making them more prone to rupture, thereby triggering
cardiovascular events (65).
Studies have demonstrated that cPLA2 can regulate autophagic flux
through multiple pathways, contributing to various cardiovascular
diseases (49,66–75)
(Fig. 3).
cPLA2 inhibits the expression and
activity of autophagy-related proteins
In the early stages, when cells are exposed to
stress, nutrient deprivation and insufficient oxygen supply,
autophagy is activated (66).
These signals include the inhibition of mTOR complex 1 (C1),
activation of AMPK and an increase in intracellular calcium ion
concentration (67,68). The Unc-51-like kinase 1 (ULK1)
complex, which consists of proteins such as ULK1, focal adhesion
kinase family interacting protein of 200 kDa, autophagy-related 13,
autophagy-related 101, as well as the Beclin-1-VPS34 complex
(including Beclin-1, VPS34 and other auxiliary proteins), are
activated (46,69). These complexes interact to regulate
downstream autophagic processes. cPLA2 hydrolyzes
phosphatidylcholine and phosphatidylethanolamine to produce free
fatty acids, which promote the accumulation of intracellular
phosphatidic acid (PA). The accumulation of PA can activate mTORC1,
which inhibits the activity of the ULK1 complex and reduces the
formation of the Beclin-1-VPS34 complex, ultimately inhibiting
autophagy. Additionally, cPLA2 can reduce the expression of
Beclin-1 by inhibiting the PI3K/Akt signaling pathway, further
suppressing autophagy (67–71).
cPLA2 reduces the formation of
autophagosome
After the initiation phase, autophagic vesicles
begin to form. These vesicles consist of a double membrane that
engulfs cellular components to be degraded. This process involves
multiple autophagy-related proteins, such as ATG9, ATG16L1 and the
ATG5-ATG12 complex (66,71). These proteins promote the formation
and expansion of autophagic vesicles through interactions and
phosphorylation. The vesicles close in a single step to form
autophagosomes. This process involves the lipidation of LC3,
wherein LC3-I is converted to LC3-II, which binds to the inner
membrane of the autophagic vesicle, promoting the formation and
closure of the autophagosome. The activation of cPLA2 can influence
the composition of membrane phospholipids, thereby affecting the
lipidation of LC3 proteins and their binding to the autophagosome
membrane. A study showed that inhibiting cPLA2 can promote the
accumulation of LC3-II (the phosphorylated form of LC3), thereby
increasing autophagosome formation (49).
cPLA2 blocks autophagy by destroying
the integrity of lysosomal membranes
After the formation of the autophagosome, it fuses
with lysosomes to form autophagic lysosomes (autolysosomes). The
components and organelles within the autophagosome are degraded by
hydrolytic enzymes in the autolysosome, facilitating the recovery
and reuse of intracellular components to meet the energy needs of
the cell and synthesize new biomolecules (57). cPLA2 is primarily responsible for
the sn-2 hydrolysis of membrane phospholipids on the cell membrane,
generating inflammatory mediators such as lysophospholipids and AA.
Lysophospholipids are a class of phosphatidylinositol metabolites
and important cell signaling molecules, including
lysophosphatidylcholine, lysophosphatidylethanolamine and
lysophosphatidylserine, among others (72). These lysophospholipids can increase
lysosomal membrane permeability by disrupting the integrity of the
autolysosome or regulating ion channels, which further disrupts the
fusion of autophagosomes with lysosomes, leading to a blockade of
autophagic flux (49,73,74).
Fusion disorders between the autophagosome and lysosome result in
impaired degradation of cellular components, promote the
transcription of inflammatory factors and exacerbate cell apoptosis
(75).
The effective role of cPLA2 in
cardiovascular diseases
The preceding section detailed the potential
mechanisms of cPLA2. Below, these mechanisms are associated with
clinical significance in cardiovascular diseases, summarizing the
research progress of cPLA2 in these conditions.
Atherosclerosis
Atherosclerosis is the underlying pathology of
cardiovascular diseases (76) and
cPLA2 plays a significant role in this process.
Amplifying inflammatory response
The formation of atherosclerosis is closely
associated with chronic low-grade inflammation (77) and cPLA2 promotes the synthesis of
inflammatory mediators, such as prostaglandins and leukotrienes.
These inflammatory mediators activate endothelial cells, induce the
recruitment of leukocytes and macrophages and initiate local
inflammatory responses. Persistent inflammation promotes lipid
accumulation in the arterial wall and accelerates plaque formation
(56).
Promoting smooth muscle cell
proliferation and migration
cPLA2 also promotes the proliferation and migration
of smooth muscle cells by regulating the release of cytokines. This
plays a critical role in the formation of atherosclerotic plaques
and vascular wall remodeling (13,56).
Blocking autophagic flux
When autophagic flux is impaired, vascular cells
(such as endothelial cells, macrophages and smooth muscle cells)
are unable to effectively clear accumulated harmful substances
(such as oxidized low-density lipoprotein and lipid droplets),
leading to enhanced inflammation, lipid deposition and plaque
formation. This further accelerates the progression of
atherosclerosis (65).
Coronary artery disease
Amplifying inflammatory response
cPLA2 can amplify the inflammatory response within
plaques by promoting the production of inflammatory mediators,
leading to plaque instability and rupture. Following plaque
rupture, thrombosis can be triggered, resulting in acute coronary
syndromes (such as myocardial infarction) (65).
Prothrombotic effect
AA derivatives produced by cPLA2 (such as
thromboxane A2) have a strong prothrombotic effect, promoting
platelet aggregation and thrombosis formation. This creates
favorable conditions for the onset of coronary heart disease
(28).
Myocardial infarction
Exacerbating inflammatory response
After myocardial infarction, the activation of cPLA2
leads to the release of AA, which further synthesizes inflammatory
mediators, such as prostaglandins and leukotrienes. These mediators
promote the infiltration of inflammatory cells (such as macrophages
and leukocytes), aggravating myocardial damage and necrosis, while
delaying myocardial repair (65).
Promoting excessive myocardial cell
apoptosis
During reperfusion therapy following acute
myocardial infarction, excessive activation of cPLA2 may lead to
excessive apoptosis of myocardial cells, exacerbating reperfusion
injury (that is, reperfusion damage), which causes further
myocardial cell death and functional loss (40).
Blocking autophagic flux
cPLA2 inhibits autophagic activity, preventing
myocardial cells from effectively clearing damaged organelles. This
leads to increased cell death, further exacerbating the damage
caused by myocardial infarction (61,62).
Heart failure
Promoting inflammatory response and cell
apoptosis
In heart failure, cPLA2 promotes myocardial cell
apoptosis and fibrosis by activating downstream inflammatory and
fibrosis pathways, leading to the deterioration of cardiac
structure and function. cPLA2 may regulate the activity of
fibroblasts, promoting collagen deposition and increasing the
stiffness of the heart, thereby worsening heart failure (37–39).
Hypertension
Endothelial injury
Increased activity of cPLA2 leads to the breakdown
of phospholipids in endothelial cell membranes, which in turn
affects the vasomotor function of blood vessels (76). Endothelial damage is a key feature
of hypertension and by altering endothelial cell function, cPLA2
may exacerbate endothelial permeability and vascular stiffness
(13), further worsening
hypertension.
Regulation of the renin-angiotensin
system
Studies suggest that cPLA2 may be involved in
regulating the action of angiotensin II (Ang II), a significant
trigger of hypertension (3,78).
By affecting this system, cPLA2 may indirectly contribute to the
elevation of blood pressure.
Valvular heart disease
Triggering inflammatory response and oxidative
stress
In valvular heart diseases (such as rheumatic heart
disease and degenerative valve disease), chronic inflammation over
time leads to the destruction and proliferation of valve tissue
(79) and the activation of cPLA2
exacerbates this process. Increased activity of cPLA2 may intensify
the generation of free radicals, thereby aggravating the
pathological damage in valve disease (1).
Affecting the function of valve
cells
Activation of cPLA2 alters the function of valve
cells (such as fibroblasts and smooth muscle cells), promoting the
proliferation, migration and collagen synthesis of these cells
(37–39). This may lead to pathological
changes such as thickening, calcification and fibrosis of the
valve, thus worsening the progression of valvular heart
disease.
Conclusion and prospects
cPLA2 plays a critical role in the onset and
progression of cardiovascular diseases, particularly in processes
such as inflammatory response, platelet activation, myocardial cell
apoptosis and autophagy. By catalyzing the hydrolysis of
phospholipids to release arachidonic acid, cPLA2 triggers a cascade
of biological reactions that promote inflammation and thrombosis,
thereby exacerbating the progression of atherosclerosis, myocardial
infarction, heart failure and other cardiovascular diseases.
Additionally, cPLA2 holds significant potential as a biomarker,
with its activity guiding early diagnosis (25), risk assessment and prognosis
evaluation in cardiovascular diseases (80,81).
Several studies have shown that inhibiting cPLA2 activity (for
example by using inhibitors such as AACOCF3) can effectively
alleviate inflammation, thrombosis, myocardial damage and other
issues associated with cardiovascular diseases (82–84).
However, its clinical application faces challenges, including lack
of specificity, unstandardized detection methods, limited
large-scale validation and the influence of various external
factors. Moreover, the development of cPLA2 inhibitors shows
promise for clinical treatment, but challenges remain in terms of
selectivity, pharmacokinetics and safety. Preclinical and clinical
trial results (for example LY3159200, developed by Eli Lilly and
Company) have demonstrated promising efficacy in experimental
settings, but further exploration is needed regarding their safety,
long-term effects and side effects (85).
In conclusion, cPLA2 inhibitors represent a novel
therapeutic approach for cardiovascular diseases. Their future
development depends on more precise drug design, comprehensive
clinical validation and thorough evaluation of long-term efficacy.
By addressing existing technological and clinical challenges,
cPLA2-targeted therapies have the potential to offer new treatment
options for cardiovascular disease patients, improve quality of
life and reduce the incidence of cardiovascular events.
Acknowledgements
Not applicable.
Funding
The present review was supported by the National Natural Science
Foundation of China (NSFC) under grant nos. 82300294 and 82202750;
Shandong Provincial Natural Science Foundation (grant no.
ZR2021QH178); Science and Technology Support Plan for Youth
Innovation of Colleges and Universities of Shandong Province of
China under grant number (grant no. 2023KJ187).
Availability of data and materials
Not applicable.
Authors' contributions
GS was involved in the conception of the study, and
the formulation and evolution of overarching study goals and aims.
XC was responsible for project administration and article revision.
WL was involved in manuscript preparation, presentation of the
information and figures, and writing the initial draft (including
substantive translation). SW contributed by preparing the
manuscript, specifically its critical review, commentary and
revision at pre-publication stages. RL performed study revisions
and improvements. DZ and XQ drew the table and diagrams and
performed revisions. JZ, ZL and MM were involved in the analysis of
the information. Data authentication is not applicable. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang S, Li B, Solomon V, Fonteh A,
Rapoport SI, Bennett DA, Arvanitakis Z, Chui HC, Sullivan PM and
Yassine HN: Calcium-dependent cytosolic phospholipase A2 activation
is implicated in neuroinflammation and oxidative stress associated
with ApoE4. Mol Neurodegener. 17:422022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin W, Zhao J, Yang E, Wang Y, Wang Q, Wu
Y, Tong F, Tan Y, Zhou J and Kang C: Neuronal STAT3/HIF-1α/PTRF
axis-mediated bioenergetic disturbance exacerbates cerebral
ischemia-reperfusion injury via PLA2G4A. Theranostics.
12:3196–3216. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song CY, Singh P, Motiwala M, Shin JS, Lew
J, Dutta SR, Gonzalez FJ, Bonventre JV and Malik KU:
2-methoxyestradiol ameliorates angiotensin II-induced hypertension
by inhibiting cytosolic phospholipase A2α activity in female mice.
Hypertension. 78:1368–1381. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jing H, Reed A, Ulanovskaya OA, Grigoleit
JS, Herbst DM, Henry CL, Li H, Barbas S, Germain J, Masuda K and
Cravatt BF: Phospholipase Cγ2 regulates endocannabinoid and
eicosanoid networks in innate immune cells. Proc Natl Acad Sci USA.
118:e21129711182021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun GY, Geng X, Teng T, Yang B, Appenteng
MK, Greenlief CM and Lee JC: Dynamic role of phospholipases A2 in
health and diseases in the central nervous system. Cells.
10:29632021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dabral D and van den Bogaart G: The roles
of phospholipase A2 in phagocytes. Front Cell Dev Biol.
9:6735022021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang HJ, Chen YT, Hu XL, Cai WT, Wang XY,
Ni WF and Zhou KL: Functions and mechanisms of cytosolic
phospholipase A2 in central nervous system trauma. Neural Regen
Res. 18:258–266. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Jones JW, Choi HMC, Sarkar C, Kane
MA, Koh EY, Lipinski MM and Wu J: cPLA2 activation contributes to
lysosomal defects leading to impairment of autophagy after spinal
cord injury. Cell Death Dis. 10:5312019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarkar C, Jones JW, Hegdekar N, Thayer JA,
Kumar A, Faden AI, Kane MA and Lipinski MM: PLA2G4A/cPLA2-mediated
lysosomal membrane damage leads to inhibition of autophagy and
neurodegeneration after brain trauma. Autophagy. 16:466–485. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayashi D, Mouchlis VD and Dennis EA: Each
phospholipase A2 type exhibits distinct selectivity toward sn-1
ester, alkyl ether, and vinyl ether phospholipids. Biochim Biophys
Acta Mol Cell Biol Lipids. 1867:1590672022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kita Y, Shindou H and Shimizu T: Cytosolic
phospholipase A2 and lysophospholipid acyltransferases. Biochim
Biophys Acta Mol Cell Biol Lipids. 1864:838–845. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Chen Y, Li F, Wu C, Cai W, Ye H,
Su H, He M, Yang L, Wang X, et al: Elamipretide alleviates
pyroptosis in traumatically injured spinal cord by inhibiting
cPLA2-induced lysosomal membrane permeabilization. J
Neuroinflammation. 20:62023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Frąk W, Wojtasińska A, Lisińska W,
Młynarska E, Franczyk B and Rysz J: Pathophysiology of
cardiovascular diseases: New insights into molecular mechanisms of
atherosclerosis, arterial hypertension, and coronary artery
disease. Biomedicines. 10:19382022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hasan S, Ghani N, Zhao X, Good J, Huang A,
Wrona HL, Liu J and Liu CJ: Dietary pyruvate targets cytosolic
phospholipase A2 to mitigate inflammation and obesity in mice.
Protein Cell. 15:661–685. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huwiler A, Feuerherm AJ, Sakem B,
Pastukhov O, Filipenko I, Nguyen T and Johansen B: The
ω3-polyunsaturated fatty acid derivatives AVX001 and AVX002
directly inhibit cytosolic phospholipase A(2) and suppress PGE(2)
formation in mesangial cells. Br J Pharmacol. 167:1691–1701. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bacher S, Meier-Soelch J, Kracht M and
Schmitz ML: Regulation of transcription factor NF-κB in its natural
habitat: The nucleus. Cells. 10:7532021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aslani M, Mortazavi-Jahromi SS and
Mirshafiey A: Cytokine storm in the pathophysiology of COVID-19:
Possible functional disturbances of miRNAs. Int Immunopharmacol.
101:1081722021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Donohoe F, Wilkinson M, Baxter E and
Brennan DJ: Mitogen-Activated protein kinase (MAPK) and
obesity-related cancer. Int J Mol Sci. 21:12412020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sahana TG and Zhang K: Mitogen-activated
protein kinase pathway in amyotrophic lateral sclerosis.
Biomedicines. 9:9692021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ye J, Zhai L, Zhang S, Zhang Y, Chen L, Hu
L, Zhang S and Ding Z: DL-3-n-butylphthalide inhibits platelet
activation via inhibition of cPLA2-mediated TXA2 synthesis and
phosphodiesterase. Platelets. 26:736–744. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Li X, Xie J, Lv C, Lian F, Zhang S,
Duan Y, Zeng Y and Piao X: Gypenoside L and Gypenoside LI Inhibit
proliferation in renal cell carcinoma via regulation of the MAPK
and arachidonic acid metabolism pathways. Front Pharmacol.
13:8206392022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lou J, Wang X, Zhang H, Yu G, Ding J, Zhu
X, Li Y, Wu Y, Xu H, Xu H, et al: Inhibition of PLA2G4E/cPLA2
promotes survival of random skin flaps by alleviating lysosomal
membrane permeabilization-induced necroptosis. Autophagy.
18:1841–1863. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng Z, Chang Y, Fan J, Ji W and Su C:
Phospholipase A2 superfamily in cancer. Cancer Lett. 497:165–177.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Zhang H, Jiang L, Cai W, Kuang J,
Geng Y, Xu H, Li Y, Yang L, Cai Y, et al: DADLE promotes motor
function recovery by inhibiting cytosolic phospholipase
A2 mediated lysosomal membrane permeabilization after
spinal cord injury. Br J Pharmacol. 181:712–734. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang Y, Hsia CW, Chiou KR, Yen TL,
Jayakumar T, Sheu JR and Huang WC: Eugenol: A potential modulator
of human platelet activation and mouse mesenteric vascular
thrombosis via an innovative cPLA2-NF-κB signaling axis.
Biomedicines. 12:16892024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Khan SA and Ilies MA: The phospholipase A2
superfamily: Structure, isozymes, catalysis, physiologic and
pathologic roles. Int J Mol Sci. 24:13532023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Elinder LS, Dumitrescu A, Larsson P, Hedin
U, Frostegård J and Claesson HE: Expression of phospholipase A2
isoforms in human normal and atherosclerotic arterial wall.
Arterioscler Thromb Vasc Biol. 17:2257–2263. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Badimon L, Vilahur G, Rocca B and Patrono
C: The key contribution of platelet and vascular arachidonic acid
metabolism to the pathophysiology of atherothrombosis. Cardiovasc
Res. 117:2001–2015. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Szczuko M, Kozioł I, Kotlęga D, Brodowski
J and Drozd A: The role of thromboxane in the course and treatment
of ischemic stroke: Review. Int J Mol Sci. 22:116442021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hong HJ, Nam GS and Nam KS: Daidzein
inhibits human platelet activation by downregulating thromboxane
A2 production and granule release, regardless of COX-1
activity. Int J Mol Sci. 24:119852023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu G, Yuan Z, Tian X, Xiong X, Guo F, Lin
Z and Qin Z: Pimpinellin inhibits collagen-induced platelet
aggregation and activation through inhibiting granule secretion and
PI3K/Akt pathway. Front Pharmacol. 12:7063632021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bertheloot D, Latz E and Franklin BS:
Necroptosis, pyroptosis and apoptosis: An intricate game of cell
death. Cell Mol Immunol. 18:1106–1121. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodríguez JP, Leiguez E, Guijas C, Lomonte
B, Gutiérrez JM, Teixeira C, Balboa MA and Balsinde J: A lipidomic
perspective of the action of group IIA secreted phospholipase A2 on
human monocytes: LIPID droplet biogenesis and activation of
cytosolic phospholipase A2α. Biomolecules. 10:8912020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Paloschi MV, Lopes JA, Boeno CN, Silva
MDS, Evangelista JR, Pontes AS, da Silva Setúbal S, Rego CMA, Néry
NM, Ferreira AA, et al: Cytosolic phospholipase A2-α participates
in lipid body formation and PGE2 release in human neutrophils
stimulated with an l-amino acid oxidase from calloselasma
rhodostoma venom. Sci Rep. 10:109762020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boi R, Ebefors K, Henricsson M, Johansson
A, Borén J and Nyström J: MO614: Modified lipid metabolism and
cytosolic phospholipase A2 activation in mesangial cells under
pro-inflammatory conditions. Nephrol Dial Transplant. May
3–2022.(Epub ahead of print). doi: 10.1093/ndt/gfac076.007, 2022.
View Article : Google Scholar
|
|
36
|
Bayır H, Anthonymuthu TS, Tyurina YY,
Patel SJ, Amoscato AA, Lamade AM, Yang Q, Vladimirov GK, Philpott
CC and Kagan VE: Achieving life through death: Redox biology of
lipid peroxidation in ferroptosis. Cell Chem Biol. 27:387–408.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li D, Chen A, Lan T, Zou Y, Zhao L, Yang
P, Qu H, Wei L, Varghese Z, Moorhead JF, et al: SCAP knockdown in
vascular smooth muscle cells alleviates atherosclerosis plaque
formation via up-regulating autophagy in ApoE-/- mice. FASEB J.
33:3437–3450. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Canty JM Jr: Myocardial injury, troponin
release, and cardiomyocyte death in brief ischemia, failure, and
ventricular remodeling. Am J Physiol Heart Circ Physiol.
323:H1–H15. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pesce M, Duda GN, Forte G, Girao H, Raya
A, Roca-Cusachs P, Sluijter JPG, Tschöpe C and Van Linthout S:
Cardiac fibroblasts and mechanosensation in heart development,
health and disease. Nat Rev Cardiol. 20:309–324. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Naik MU, Patel P, Derstine R, Turaga R,
Chen X, Golla K, Neeves KB, Ichijo H and Naik UP: Ask1 regulates
murine platelet granule secretion, thromboxane A2 generation, and
thrombus formation. Blood. 129:1197–1209. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yue J and López JM: Understanding MAPK
signaling pathways in apoptosis. Int J Mol Sci. 21:23462020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xuan C, Jin C, Jin Z and Chi Y: The
protective effects of glutamine against bronchopulmonary dysplasia
are associated with MKP-1/MAPK/cPLA2 signalingmediated NF-kappaB
pathway. Gen Physiol Biophys. 42:229–239. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Davidovich P, Higgins CA, Najda Z, Longley
DB and Martin SJ: cFLIPL acts as a suppressor of TRAIL- and
Fas-initiated inflammation by inhibiting assembly of
caspase-8/FADD/RIPK1 NF-κB-activating complexes. Cell Rep.
42:1134762023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mustafa M, Ahmad R, Tantry IQ, Ahmad W,
Siddiqui S, Alam M, Abbas K, Moinuddin, Hassan MI, Habib S and
Islam S: Apoptosis: A comprehensive overview of signaling pathways,
morphological changes, and physiological significance and
therapeutic implications. Cells. 13:18382024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Whitaker RH and Cook JG: Stress relief
techniques: p38 MAPK determines the balance of cell cycle and
apoptosis pathways. Biomolecules. 11:14442021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang A, Jiang H, Liu Y, Chen J, Zhou X,
Zhao C, Chen X and Lin M: Rhein induces liver cancer cells
apoptosis via activating ROS-dependent JNK/Jun/caspase-3 signaling
pathway. J Cancer. 11:500–507. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ke Z, Lu J, Zhu J, Yang Z, Jin Z and Yuan
L: Down-regulation of lincRNA-EPS regulates apoptosis and autophagy
in BCG-infected RAW264.7 macrophages via JNK/MAPK signaling
pathway. Infect Genet Evol. 77:1040772020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng N, Li H, Wang X, Zhao Z and Shan D:
Oxidative stress-induced cardiomyocyte apoptosis is associated with
dysregulated Akt/p53 signaling pathway. J Recept Signal Transduct
Res. 40:599–604. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zou H and Liu G: Inhibition of endoplasmic
reticulum stress through activation of MAPK/ERK signaling pathway
attenuates hypoxia-mediated cardiomyocyte damage. J Recept Signal
Transduct Res. 41:532–537. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xu F, Na L, Li Y and Chen L: Roles of the
PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and
tumours. Cell Biosci. 11:1572020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Qin W, Cao L and Massey IY: Role of
PI3K/Akt signaling pathway in cardiac fibrosis. Mol Cell Biochem.
476:4045–4059. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Peng Y, Wang Y, Zhou C, Mei W and Zeng C:
PI3K/Akt/mTOR pathway and its role in cancer therapeutics: Are we
making headway? Front Oncol. 12:8191282022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen J, Tang H, Hay N, Xu J and Ye RD: Akt
isoforms differentially regulate neutrophil functions. Blood.
115:4237–4246. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lin WC, Chuang YC, Chang YS, Lai MD, Teng
YN, Su IJ, Wang CC, Lee KH and Hung JH: Endoplasmic reticulum
stress stimulates p53 expression through NF-κB activation. PLoS
One. 7:e391202012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cheng W, Cui C, Liu G, Ye C, Shao F,
Bagchi AK, Mehta JL and Wang X: NF-κB, A potential therapeutic
target in cardiovascular diseases. Cardiovasc Drugs Ther.
37:571–584. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang XW, Lv XX, Zhou JC, Jin CC, Qiao LY
and Hu ZW: Autophagic flux detection: Significance and methods
involved. Adv Exp Med Biol. 1208:131–173. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mi S, Huang F, Jiao M, Qian Z, Han M, Miao
Z and Zhan H: Inhibition of MEG3 ameliorates cardiomyocyte
apoptosis and autophagy by regulating the expression of
miRNA-129-5p in a mouse model of heart failure. Redox Rep.
28:22246072023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu X, Liu Z, Yu XY, Xu S and Luo J:
Autophagy and cardiac diseases: Therapeutic potential of natural
products. Med Res Rev. 41:314–341. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Che Y, Wang Z, Yuan Y, Zhou H, Wu H, Wang
S and Tang Q: By restoring autophagic flux and improving
mitochondrial function, corosolic acid protects against Dox-induced
cardiotoxicity. Cell Biol Toxicol. 38:451–467. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang Q, Su H and Liu J: Protective effect
of natural medicinal plants on cardiomyocyte injury in heart
failure: Targeting the dysregulation of mitochondrial homeostasis
and mitophagy. Oxid Med Cell Longev. 2022:36170862022.PubMed/NCBI
|
|
63
|
Gao J, Chen X, Shan C, Wang Y, Li P and
Shao K: Autophagy in cardiovascular diseases: Role of noncoding
RNAs. Mol Ther Nucleic Acids. 23:101–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang L, Wang J, Cretoiu D, Li G and Xiao
J: Exercise-mediated regulation of autophagy in the cardiovascular
system. J Sport Health Sci. 9:203–210. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Miao J, Zang X, Cui X and Zhang J:
Autophagy, hyperlipidemia, and atherosclerosis. Adv Exp Med Biol.
1207:237–264. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yun HR, Jo YH, Kim J, Shin Y, Kim SS and
Choi TG: Roles of autophagy in oxidative stress. Int J Mol Sci.
21:32892020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Frias MA, Hatipoglu A and Foster DA:
Regulation of mTOR by phosphatidic acid. Trends Endocrinol Metab.
34:170–180. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sukumaran P, Da Conceicao VN, Sun Y,
Ahamad N, Saraiva LR, Selvaraj S and Singh BB: Calcium signaling
regulates autophagy and apoptosis. Cells. 10:21252021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Y, Yang Q, Chen S, Li Z and Fu L:
Targeting VPS34 in autophagy: An update on pharmacological
small-molecule compounds. Eur J Med Chem. 256:1154672023.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Foster KG and Fingar DC: Mammalian target
of rapamycin (mTOR): Conducting the cellular signaling symphony. J
Biol Chem. 285:14071–14077. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Gao G, Chen W, Yan M, Liu J, Luo H, Wang C
and Yang P: Rapamycin regulates the balance between cardiomyocyte
apoptosis and autophagy in chronic heart failure by inhibiting mTOR
signaling. Int J Mol Med. 45:195–209. 2020.PubMed/NCBI
|
|
72
|
Jiang S, Yang H and Li M: Emerging roles
of lysophosphatidic acid in macrophages and inflammatory diseases.
Int J Mol Sci. 24:125242023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang ML, Zhang YJ, He DL, Li T, Zhao MM
and Zhao LM: Inhibition of PLA2G4A attenuated valproic acid-induced
lysosomal membrane permeabilization and restored impaired
autophagic flux: Implications for hepatotoxicity. Biochem
Pharmacol. 227:1164382024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yang HL, Lai ZZ, Shi JW, Zhou WJ, Mei J,
Ye JF, Zhang T, Wang J, Zhao JY, Li DJ and Li MQ: A defective
lysophosphatidic acid-autophagy axis increases miscarriage risk by
restricting decidual macrophage residence. Autophagy. 18:2459–2480.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ballabio A and Bonifacino JS: Lysosomes as
dynamic regulators of cell and organismal homeostasis. Nat Rev Mol
Cell Biol. 21:101–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kotlyarov S: Immune function of
endothelial cells: Evolutionary aspects, molecular biology and role
in atherogenesis. Int J Mol Sci. 23:97702022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gusev E and Sarapultsev A: Atherosclerosis
and inflammation: Insights from the theory of general pathological
processes. Int J Mol Sci. 24:79102023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Singh P, Song CY, Dutta SR, Pingili A,
Shin JS, Gonzalez FJ, Bonventre JV and Malik KU:
6β-Hydroxytestosterone promotes angiotensin II-induced hypertension
via enhanced cytosolic phospholipase A2α activity. Hypertension.
78:1053–1066. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Passos LSA, Nunes MCP and Aikawa E:
Rheumatic heart valve disease pathophysiology and underlying
mechanisms. Front Cardiovasc Med. 7:6127162021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang H, Gao Y, Wu D and Zhang D: The
relationship of lipoprotein-associated phospholipase A2 activity
with the seriousness of coronary artery disease. BMC Cardiovasc
Disord. 20:2952020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Verdoia M, Rolla R, Gioscia R, Rognoni A
and De Luca G; Novara Atherosclerosis Study Group (NAS), :
Lipoprotein associated- phospholipase A2 in STEMI vs. NSTE-ACS
patients: A marker of cardiovascular atherosclerotic risk rather
than thrombosis. J Thromb Thrombolysis. 56:37–44. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yigit E, Deger O, Korkmaz K, Yigit MH,
Uydu HA, Mercantepe T and Demir S: Propolis reduces inflammation
and dyslipidemia caused by high-cholesterol diet in mice by
lowering ADAM10/17 activities. Nutrients. 16:18612024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ashcroft FJ, Mahammad N, Flatekvål HM,
Feuerherm AJ and Johansen B: cPLA2α enzyme inhibition attenuates
inflammation and keratinocyte proliferation. Biomolecules.
10:14022020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Schanstra JP, Luong TTD, Makridakis M, Van
Linthout S, Lygirou V, Latosinska A, Alesutan I, Boehme B, Schelski
N, Von Lewinski D, et al: Systems biology identifies cytosolic PLA2
as a target in vascular calcification treatment. JCI Insight.
4:e1256382019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Huang JP, Cheng ML, Wang CH, Huang SS,
Hsieh PS, Chang CC, Kuo CY, Chen KH and Hung LM: Therapeutic
potential of cPLA2 inhibitor to counteract dilated-cardiomyopathy
in cholesterol-treated H9C2 cardiomyocyte and MUNO rat. Pharmacol
Res. 160:1052012020. View Article : Google Scholar : PubMed/NCBI
|