Ischemia-reperfusion injury (IRI) is a clinical
phenomenon encountered in conditions such as myocardial infarction,
stroke, organ transplantation and surgical intervention. In these
contexts, tissues or organs undergo ischemia due to compromised
blood supply, followed by irreversible damage following reperfusion
(1). The pathophysiology of IRI
typically unfolds in several key stages: i) During ischemia, tissue
is deprived of oxygen, leading to hypoxia and the accumulation of
metabolites; this results in a notable decline in intracellular ATP
levels, impairing cellular function and culminating in cell death;
ii) in the reperfusion phase, restoration of blood flow
re-establishes oxygenation but the sudden influx of oxygen may
initiate a cascade of detrimental reactions, including oxidative
stress and release of inflammatory mediators. These responses
exacerbate cell injury, intensifying tissue necrosis and functional
deterioration (2). Studies on the
role of aldehyde dehydrogenase (ALDH)2 in IRI are summarized in
Table I (3–25).
Ferroptosis is a more recently identified form of
regulated cell death (RCD), characterized by intracellular iron
accumulation and subsequent oxidative stress, which induces both
apoptosis and necrosis (32). IRI
triggers release and accumulation of intracellular iron, amplifying
cellular damage through oxidative stress and promoting inflammatory
responses (32). While the
mechanisms of ferroptosis in IRI are complex, its key role in the
injury cascade has been well-established (32,33).
As such, targeting ferroptosis regulation has emerged as a
promising strategy for mitigating IRI (33).
IRI is a multifaceted process that involves a range
of cellular and molecular alterations. Despite application of
certain therapeutic interventions in clinical practice, such as
antioxidants (vitamin E, vitamin C and N-acetylcysteine),
anti-inflammatory (corticosteroids, non-steroidal anti-inflammatory
drugs), Calcium Channel Blockers (verapamil, dantrolene) effective
treatment for IRI remains challenging. Thus, the identification of
novel therapeutic targets and strategies is essential. Recent
research underscores the importance of RCD pathways, particularly
ferroptosis, in the pathogenesis of IRI (2,34).
However, the mechanisms that regulate ferroptosis during IRI remain
poorly understood. The present review aimed to summarize the roles
of ALDH2 and ferroptosis in the context of IRI. ALDH2 has been
proposed as a key regulator of ferroptosis. By detoxifying reactive
aldehydes and preventing lipid peroxidation (LPO), ALDH2 may
influence ferroptosis, thereby modulating severity of IRI (35,36).
Additionally, ALDH2 regulates other forms of programmed cell death
(20,37). The present aimed to review
summarize these pathways to identify potential commonalities in
ALDH2 regulation of ferroptosis and the mechanisms underlying
cellular damage and inflammation to provide insight into their
potential therapeutic applications. Through a comprehensive
examination of ALDH2 and ferroptosis, the present review aims to
provide a theoretical framework for development of innovative
treatments and pharmacotherapies to improve clinical outcomes.
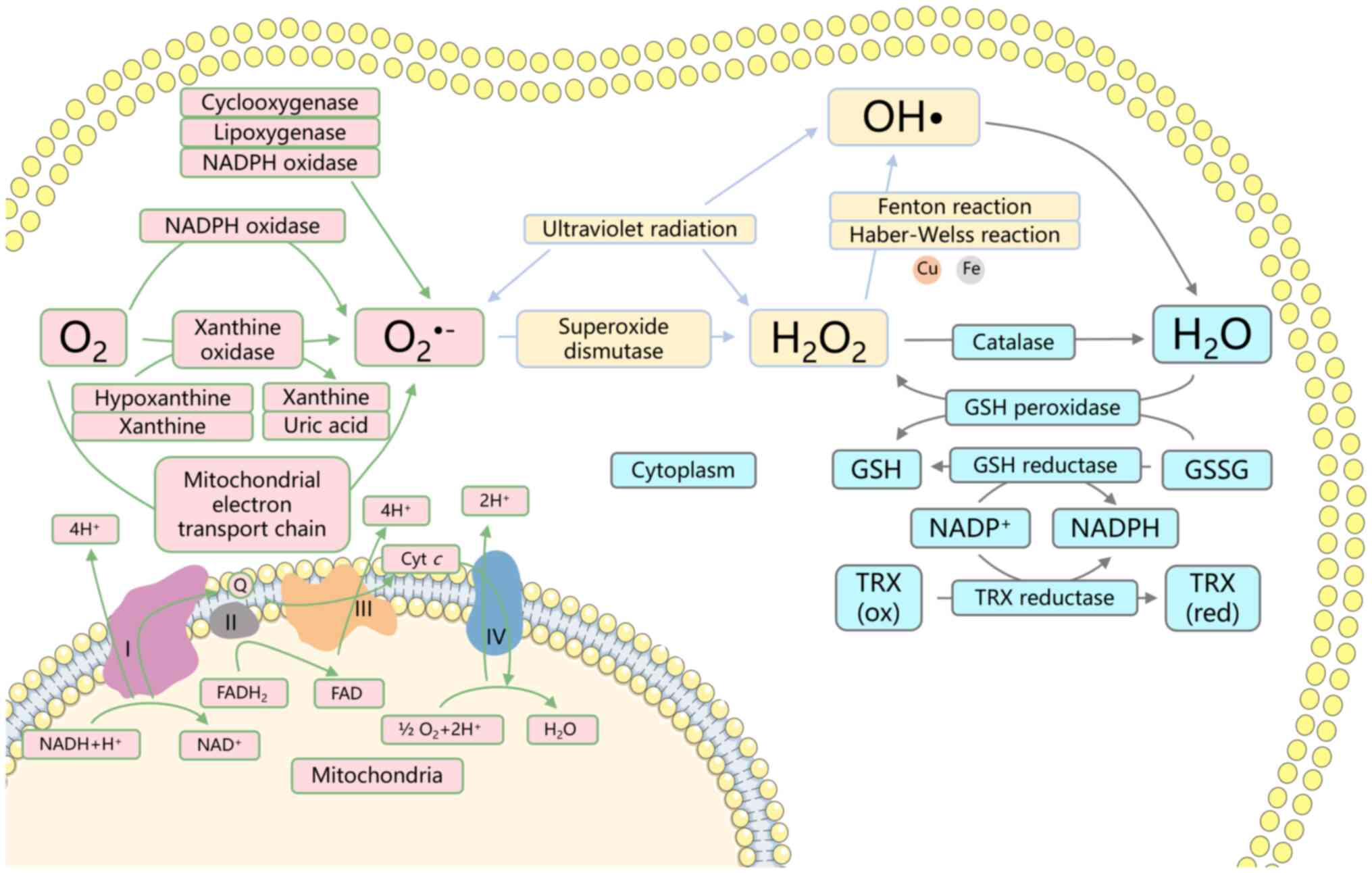
ROS are highly reactive molecules, such as
superoxide anion, hydrogen peroxide (H2O2)
and hydroxyl radicals (OH•), produced as byproducts of normal cell
metabolism (38) (Fig. 1). However, excessive ROS
accumulation can lead to oxidative stress and damage to proteins,
lipids and DNA, which results in cellular injury and potentially
cell death. ALDH2 functions as a potent scavenger of ROS (26). By degrading toxic aldehydes, ALDH2
decreases intracellular aldehyde levels, mitigating their harmful
effects on cellular integrity (39). Aldehydes, with strong oxidative
potential, bind and oxidize cellular macromolecules, including
proteins and DNA, thereby inducing cell damage. Additionally,
during the conversion of acetaldehyde to acetic acid, ALDH2
catalyzes the reduction of NAD+ to NADH (40). NADH serves as a substrate for
mitochondrial respiratory chain complex I, promoting oxidative
phosphorylation and enhancing the efficiency of redox reactions,
which limits ROS production. Furthermore, elevated ROS levels can
influence ALDH2 activity and stability (41,42),
establishing a feedback loop that modulates ALDH2 function.
In IRI, evidence highlights the role of ALDH2 in
mitigating ROS production and protecting tissues and organs
(18,20). In rat diabetic myocardial IRI and
H9C2 cardiomyocyte hypoxia-reoxygenation models, Tan et al
(18) observed that ALDH2
upregulation decreases mitochondrial ROS levels. This facilitates
maintenance of mitochondrial dynamics, preserving the balance
between fusion and fission and stabilizing mitochondrial
morphology. Xu et al (20)
revealed that ALDH2 activation promotes autophagy by increasing the
phosphorylation of Beclin-1 at Ser90. This autophagy activation
maintains cellular and tissue homeostasis by reducing ROS levels
and clearing damaged organelles. These findings underscore the
multifaceted protective mechanisms mediated by ALDH2 in IRI, from
ROS reduction to preservation of mitochondrial dynamics and
promotion of autophagy. These insights contribute to understanding
of the complex interactions between ALDH2 and IRI, offering a
foundation for development of targeted therapeutic interventions
aimed at reducing tissue damage and improving clinical
outcomes.
LPO is a key feature of IRI. Elevated ROS levels
within cells directly target lipids, with unchecked oxidative
stress disrupting the balance between ROS production and clearance,
leading to accumulation of endogenous ROS (43). Of ROS species involved in LPO,
hydroxyl and hydroperoxyl radicals are primary contributors. These
radicals attack the carbon-carbon double bonds of polyunsaturated
phospholipids in membranes, triggering a cascade of oxidation that
compromises membrane integrity and impairs membrane functionality
(44–46).
Membrane lipids, primarily composed of polar
glycerophospholipids, are susceptible to oxidative damage. Cell
membranes contain notable amounts of polyunsaturated lipids, which
are vulnerable during IR, a phase in which mitochondria become the
principal site of ROS generation (47). ROS interact with polyunsaturated
phospholipids in mitochondrial membranes, forming lipid
hydroperoxides. Secondary reactions produce >200 types of highly
reactive lipid-derived aldehydes (LDAs), such as 4-HNE, MDA,
4-oxononenal, acrolein, crotonaldehyde and methylglyoxal (47). These LDAs not only serve as ROS
byproducts but also further stimulate ROS production in
mitochondria, intensifying tissue damage through a positive
feedback loop. Consequently, excess aldehydes and ROS diffuse from
mitochondria to other cell compartments, precipitating protein
dysfunction, DNA damage and gene mutations, contributing to a range
of diseases, including cardiovascular conditions, cancer, diabetes,
osteoporosis and neurodegenerative disorder (48).
As markers of oxidative stress, 4-HNE and MDA are
among the most potent reactive aldehydes (49). ALDH2 serves a key role in their
detoxification by metabolizing MDA to malonic acid or acetaldehyde
and converting 4-HNE to 4-hydroxy-2-nonenic acid. By reducing the
accumulation of these toxic aldehydes, ALDH2 contributes to
mitigating oxidative stress (49).
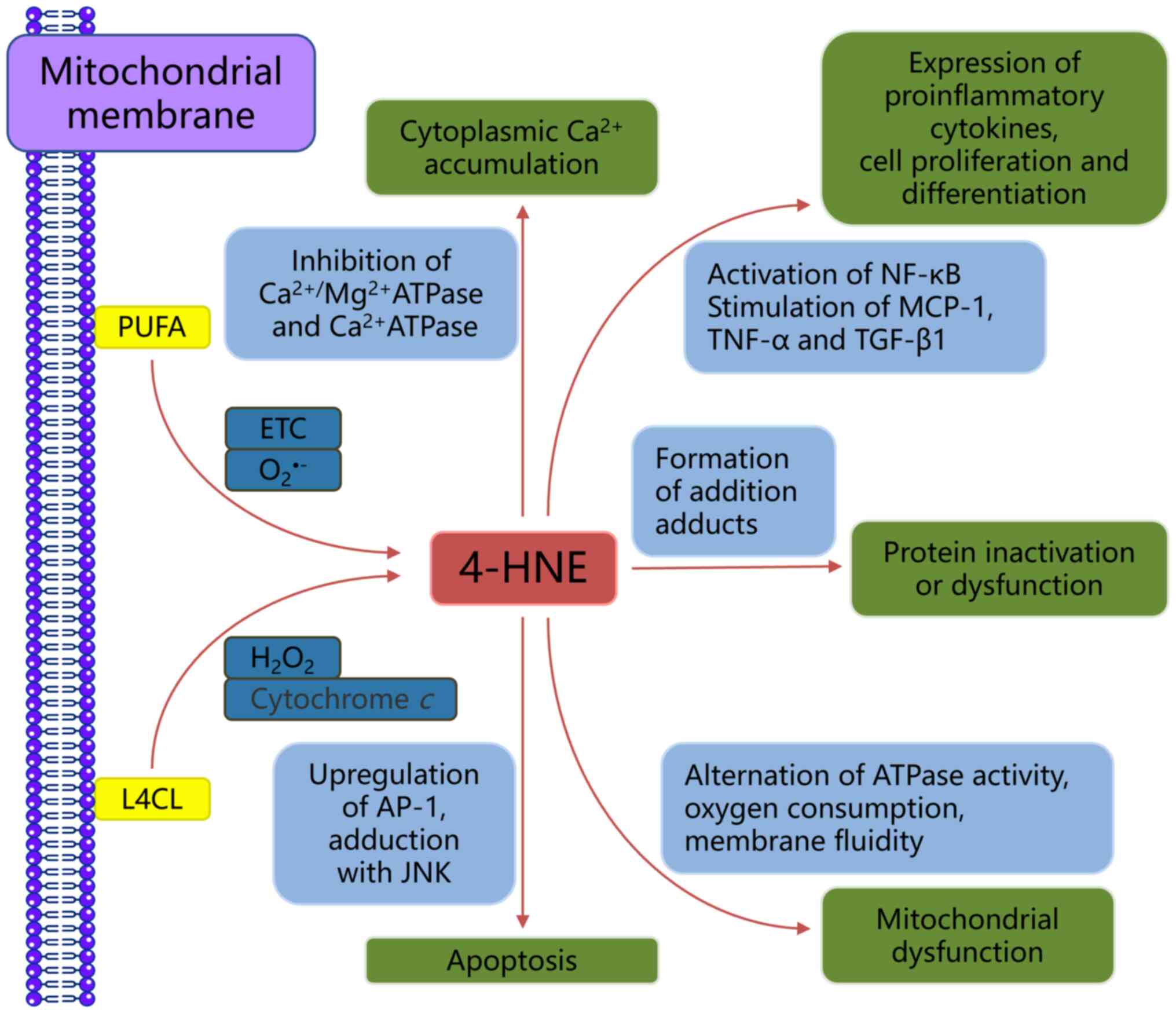
As aforementioned, 4-HNE results from LPO, with ω-6
polyunsaturated fatty acids (PUFAs) such as linoleic, γ-linolenic
and arachidonic acid (AA) serving as precursors under oxidative
stress mediated by ROS (50)
(Fig. 2). 4-HNE primarily targets
reduction-associated signaling molecules and upstream regulatory
proteins, including thioredoxin (an intracellular ROS scavenger)
and glutathione (GSH) (51).
Additionally, 4-HNE affects protein post-translational
modifications and gene expression: It upregulates the expression of
transcription factors such as NF-κB and activating protein-1, which
regulate genes involved in cell proliferation and differentiation
(52). Moreover, 4-HNE activates
transcription factor nuclear factor erythroid 2-related factor 2
(Nrf2), which transactivates antioxidant response elements and
enhances the expression of genes that are key for cell antioxidant
defense and modulation of oxidative stress. Notable genes regulated
by Nrf2 include hemeoxygenase-1 (HO-1), ALDH, glutathione
S-transferase, multidrug resistance protein, aldo-keto reductase,
nicotinamide adenine dinucleotide phosphate: quinone oxidoreductase
(NQO1) and glutamate-cysteine ligase (53).
4-HNE is implicated in a range of oxidative
stress-associated diseases, including neurodegenerative disorders,
macular degeneration, cardiovascular disease, atherosclerosis,
metabolic syndrome and cancer. In these pathological conditions,
4-HNE transcends its role as an oxidative stress marker to become a
key pathogenic factor (54).
Accumulation of 4-HNE has been detected in various types of tissue
during IRI such as heart, brain and intestine. ALDH2 has a key role
in neutralizing 4-HNE; loss of ALDH2 function exacerbates
myocardial cell ferroptosis induced by iron overload, whereas ALDH2
activation mitigates ferroptosis (35). Furthermore, compounds such as
isorhapontigenin have shown efficacy in decreasing oxidative damage
in brain IRI through the PKCε/Nrf2/HO-1 pathway, markedly lowering
ROS generation and 4-HNE and 8-hydroxydeoxyguanosine levels
(55). Similar protective effects
have been observed in intestinal IRI, where administration of
corilagin, an IRI protectant, reduces 4-HNE levels and ferroptosis
(56). 4-HNE can react with
various cellular components, forming adducts that contribute to
tissue damage. Numerous studies have revealed the protective role
of ALDH2 in IRI by effectively removing 4-HNE (18,57).
Transgenic mice with impaired ALDH2 2*2 activity show elevated
4-HNE and GSH levels, along with decreased resistance to acute
oxidative stress induced by IR (57). Additionally, recent evidence has
highlighted the ability of ALDH2 to improve cardiac hemodynamic
parameters and reduce myocardial injury by lowering 4-HNE levels
and inhibiting mitochondrial permeability transition pore opening
(18). In liver IRI,
supplementation of ALDH2 activator Alda-1 reduces 4-HNE
accumulation, reverses mitochondrial damage and decreases
hepatocyte apoptosis (11). These
findings underscore the key role of ALDH2 in mitigating harmful
effects of 4-HNE accumulation in IRI in various tissues and
organs.
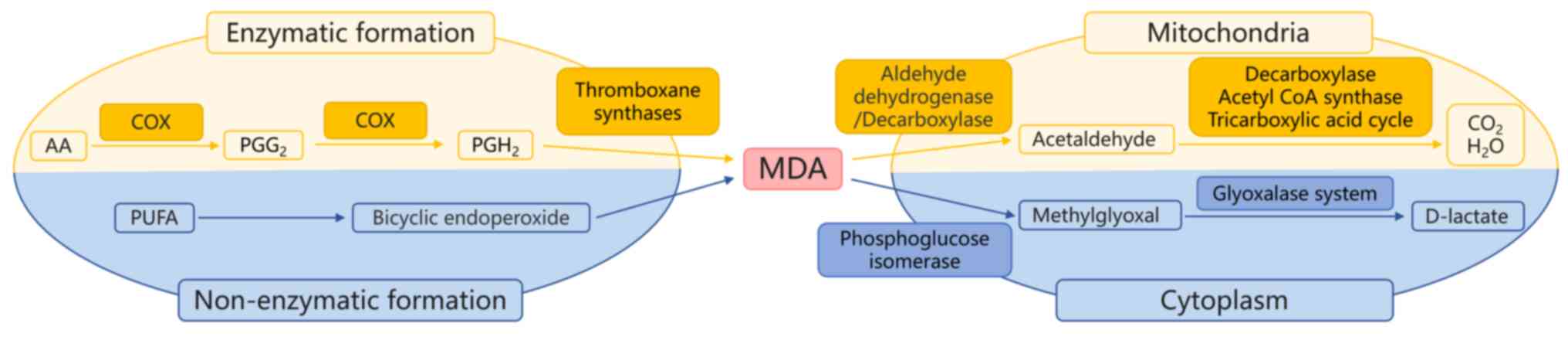
PUFAs, including AA and docosahexaenoic acid, serve
as precursors for MDA generation. PUFAs with ≥2
methylene-interrupted double bonds are susceptible to oxidative
degradation, resulting in MDA production (58) (Fig.
3). Thiobarbituric acid (TBA) assay, initially developed by
food chemists to assess the rancidity of Ω-3 and −6 fatty acids, is
commonly used for the colorimetric or fluorometric determination of
MDA and MDA-like substances (59).
However, TBA reacting substances test lacks specificity. Treatment
of biological samples with TBA may generate complexes with similar
colors to the TBA-MDA complex, potentially confounding
quantification of MDA. Furthermore, MDA or MDA-like substances are
derived from various precursors produced during LPO, including
oxidized lipids, 2-alkenals and 2,4-alkadienals (58).
While MDA is the primary aldehyde generated during
LPO, production of 4-HNE constitutes only 10% of MDA (60). Despite this, the role of MDA as a
signaling molecule and gene expression regulator remains is
understudied compared with 4-HNE. MDA, however, has gained
attention as a biomarker in various types of disease, including
cancer, cardiovascular diseases and neurodegenerative disorders,
with its levels in blood, urine and exhaled breath condensate used
for disease detection (61,62).
MDA-modified low-density lipoprotein (LDL) particles serve a key
role in atherosclerosis pathogenesis (63). These modified LDL particles are
efficiently recognized and internalized by vascular wall
macrophages by scavenger receptors, leading to accumulation in
lipid vesicles. The subsequent engulfment of MDA-modified LDLs by
macrophages results in foam cell formation, initiating lipid
deposition in the vascular wall, which represents early
atherosclerotic lesions (63).
Moreover, MDA conjugates with autologous biomolecules to generate
neo-self epitopes, which interact with the innate immune system.
Effects of various MDA epitopes are mediated via protein kinase C
pathways, including PI3K, Src kinase, phospholipase C/inositol
trisphosphate (IP3), Erk 1/2, spleen tyrosine kinase and NF-κB
(64). These findings underscore
the multifaceted role of MDA in disease pathogenesis and immune
modulation, positioning it as a potential therapeutic target in
various pathological conditions.
MDA undergoes enzymatic metabolism, with the ALDH
family in mitochondria serving a central role in its degradation.
Initially, MDA is oxidized to acetaldehyde by ALDH, which is
further oxidized to acetate. Acetate is then converted to carbon
dioxide and water. Alternatively, MDA metabolism occurs via
GSH-mediated pathways, where phosphoglucose isomerase uses GSH as a
cofactor to convert MDA to methylglyoxal (MG) in the cytoplasm. MG
is metabolized to D-lactate by the glyoxalase system enzymes
(65) (Fig. 3). In the context of IRI, elevated
oxidative stress in cells triggers considerable MDA production,
contributing to tissue and organ dysfunction. Modulating up- and
downstream molecular pathways to attenuate MDA production can
markedly alleviate IRI severity (66,67).
In cerebral IRI, reducing MDA production and inflammatory mediators
alleviates brain tissue damage by inhibiting phosphorylation and
nuclear translocation of NF-κB (66). Similarly, in the liver, the
Nrf2/solute carrier family 7 member 11 (SLC7A11)-HO-1 axis
regulates MDA production and inhibits ferroptosis (67). The role of MDA in cardiac IRI has
been extensively studied, revealing that inhibiting ROS production
and MDA levels and enhancing superoxide dismutase activity
mitigates oxidative stress in vitro (5,68).
These interventions also inhibit ferroptosis and reduce apoptotic
protein levels (68). The cell MDA
levels are associated with ALDH2 activity, with animal models of
acute lung injury (69), cardiac
reperfusion (5) and liver IR
(11) demonstrating a negative
regulatory association between ALDH2 activity and MDA levels.
4-HNE and MDA are among the most potent and
extensively studied aldehydes in IRI (18,57,60,66,67).
ALDH2 exhibits varying sensitivity to LPO-derived aldehydes, with
the highest efficacy in oxidizing 4-HNE, acrolein and MDA (70). To the best of our knowledge,
however, research on ALDH2 and toxic aldehydes predominantly
focuses on the heart (5,18,35,68),
with limited studies on other organs such as the brain, intestine,
kidney and liver. Although the pathophysiological mechanisms of IRI
are largely similar across different types of tissue, structural
and functional differences in various organs result in distinct
responses to oxidative stress. While several studies document
decreased ALDH2 activity during oxidative stress (1,26,31),
ALDH2 activity remains stable (71). Thus, investigating ALDH2
alterations in different tissues to enable the development of
targeted therapeutic strategies is key. By exploring the
differential responses of ALDH2 to oxidative stress in different
types of tissues, valuable insights can be gained into
tissue-specific vulnerability, leading to tailored interventions
aimed at mitigating IRI. This underscores the need for continued
research to unravel the complex interactions between ALDH2 and
oxidative stress in diverse physiological contexts.
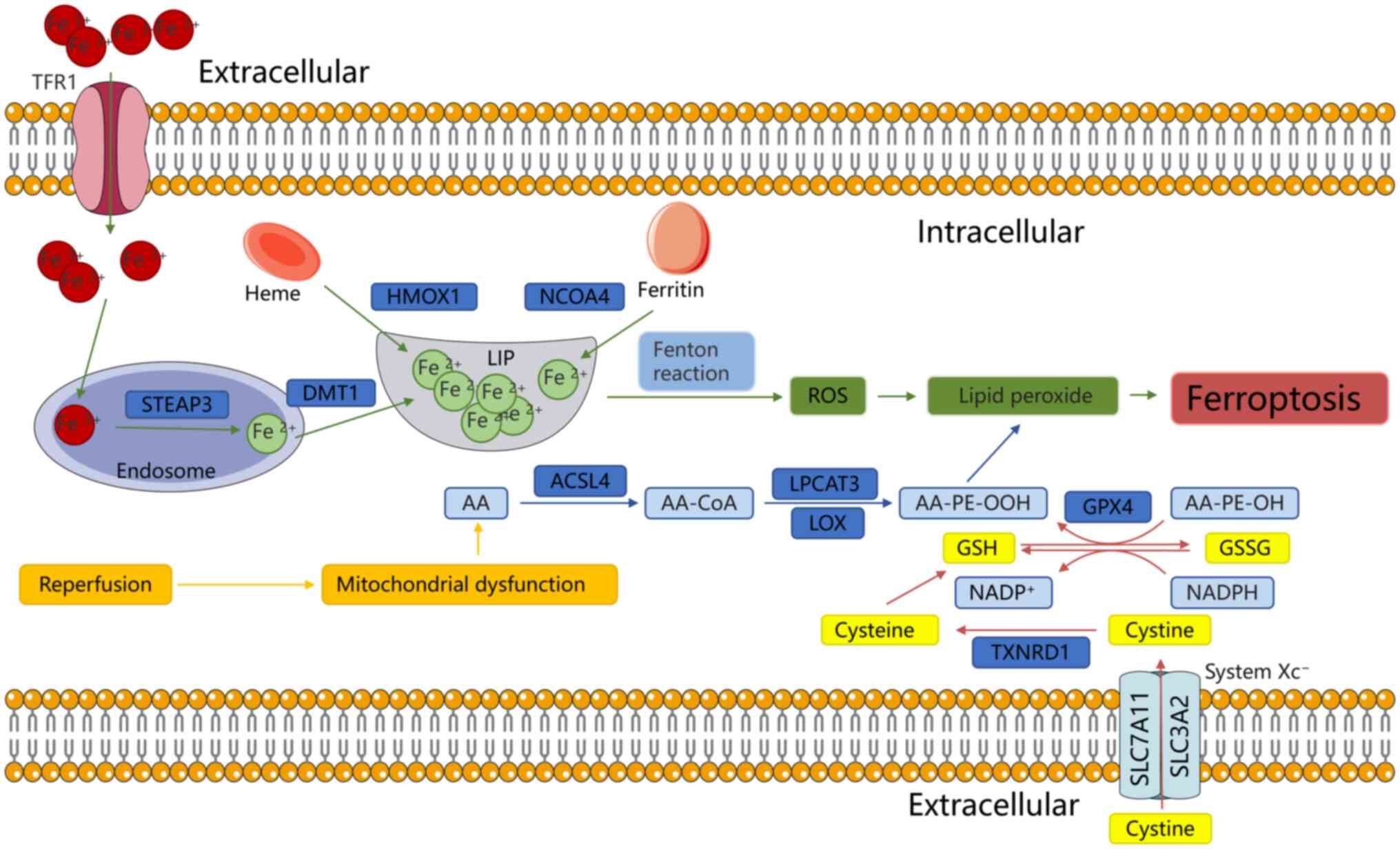
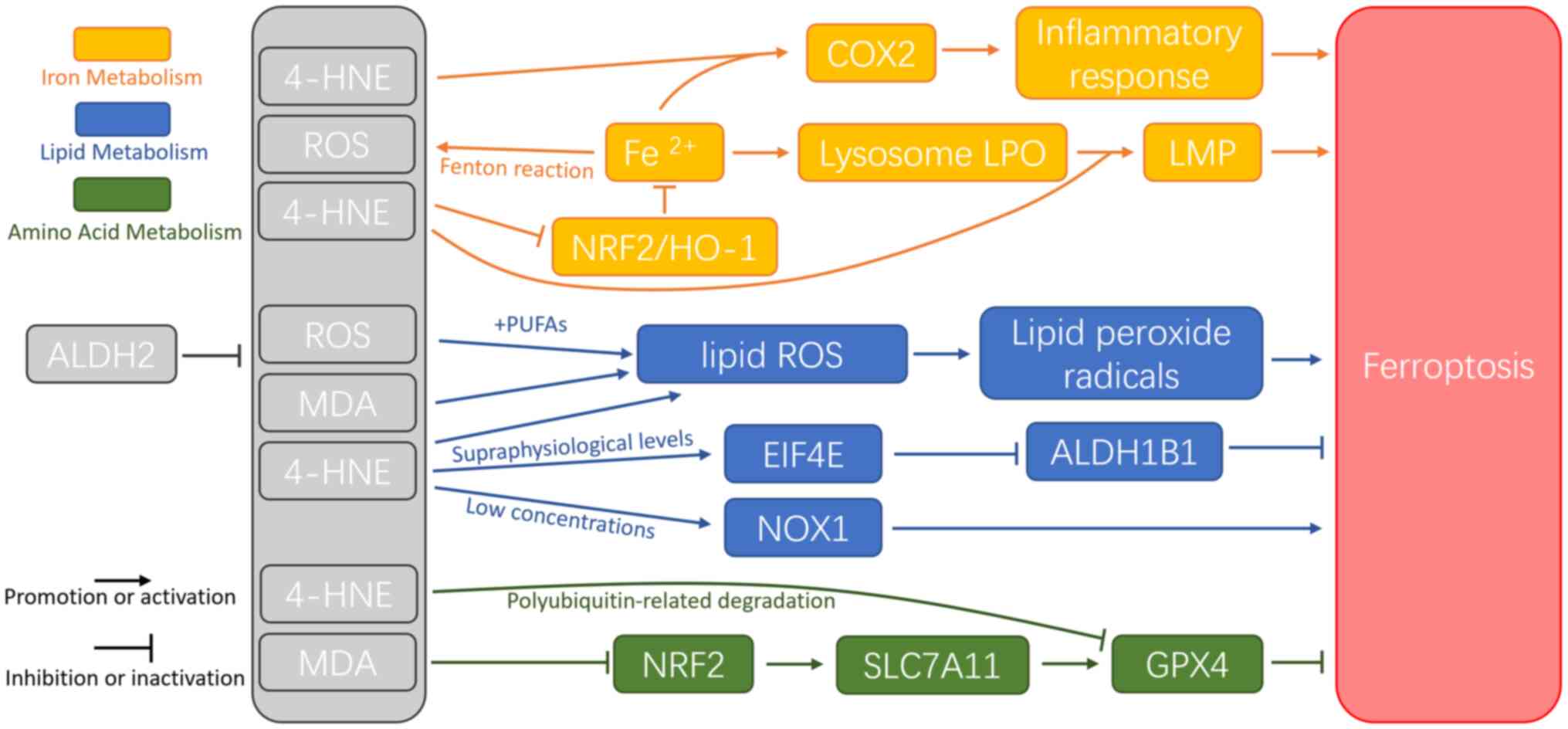
Ferroptosis is a more recently identified
iron-dependent form of RCD, characterized by excessive ROS
accumulation and LPO (32). During
IRI, ferroptosis predominantly occurs during the reperfusion phase
rather than the ischemic phase (72), marked by GSH depletion and
inactivation of GSH peroxidase 4 (GPX4) (32). Ferroptosis is the primary form of
cell death during IR in various organs, including the brain, kidney
and heart (73). The mechanisms
underlying ferroptosis involve numerous interconnected pathways,
including lipid, iron and amino acid metabolism (74) (Fig.
4).
Lipid metabolism serves a key role in ferroptosis
during IRI. LPO is initiated by the oxidation of PUFAs,
particularly AA-containing phosphatidylethanolamine (AA-PE)
(75). ROS generated by the Fenton
reaction contribute to LPO (76).
PUFAs exhibit a high affinity for ROS, such as OH• and
H2O2, which remove hydrogen atoms from PUFAs,
generating lipid ROS. These lipid radicals are further oxidized to
lipid peroxide radicals (LOO−) or AA-OOH-PE (77). When the reduction system fails to
neutralize excess of LOO•, ferroptosis is triggered (78,79).
In mice, ischemia induces arachidonic acid 15-lipoxygenase to
oxidize AA-PE into oxidized phosphatidylethanolamines, which,
following reperfusion, initiate LPO and promote ferroptosis
(72). Lipoxygenases (LOXs)
catalyze generation of ROS (34)
and oxidation of AA-PE to AA-OOH-PE (80); while LOXs are considered to be
iron-dependent enzymes (81),
another study has reported iron-oxidizing phospholipids independent
of LOXs (82). In IRI, expression
of LOX increases with extended reperfusion, reaching a peak at 5 h
post-reperfusion (83). The
accumulation of toxic aldehydes, such as 4-HNE and MDA, also serve
a key role in ferroptosis (35).
Biomarkers such as MDA levels, the glutathione disulfide (GSSG)
ratio and Fe2+ and ROS levels are used to assess
ferroptosis in cells (84,85). Additionally, 4-HNE impedes the
accumulation of ALDH1 family member B1 by activating eukaryotic
initiation factor 4E and induces ferroptosis by activating NADPH
oxidase 1 (86).
Iron serves a dual role in ferroptosis: It promotes
ROS accumulation via the Fenton reaction and serves as a key
element in enzymes such as LOXs. The Fenton reaction accelerates
production of OH•; excessive OH• exacerbates LPO, yielding 4-HNE
and activating cyclooxygenase 2 (COX2). COX2, a pro-inflammatory
enzyme, triggers cellular inflammation and may be released into the
extracellular space by vesicles (87,88).
Furthermore, 4-HNE induces mitochondrial dysfunction by binding to
mitochondrial proteins, leading to electron leakage and increased
ROS production. This activates NRF2-mediated antioxidant responses
(88).
Iron overload serves a key role in ferroptosis
through several mechanisms. i) HO-1 catalyzes release of
Fe2+ from hemoglobin, contributing to iron overload
(89); ii) nuclear receptor
coactivator 4 (NCOA4) mediates ferritin degradation by autophagy
(ferritinophagy), releasing free iron (90) iii) transferrin receptor protein 1
(TFR1) induces ferroptosis during IRI, with p53 promoting TFR1
upregulation through deubiquitination of ubiquitin-specific
protease 7 (91). Both
ferritinophagy and TFR1 are considered key regulators of iron
accumulation (92). This iron is
released into the labile iron pool in the cytoplasm. During IR,
ferritinophagy induces intracellular iron overload, leading to
ferroptosis. Targeting ferritinophagy with inhibitors notably
decreases organ damage (93,94).
Furthermore, ubiquitination and proteasomal degradation of TFR1
counteract abnormal iron accumulation and ferroptosis, mitigating
acute liver injury (95). During
ferroptosis, endocytosis of extracellular ferritin increases
Fe2+ levels in lysosomes. The release of free
Fe2+ from lysosomes accelerates ROS formation and
accumulation of lysosomal LPO. This generates 4-HNE and HNE
adducts, leading to lysosomal membrane permeabilization. Detection
of HNE adducts in the cytoplasm suggests that LPO diffuses from the
lysosome into the cytosol (96).
Numerous studies have highlighted the involvement of
cell death modalities in IRI, including apoptosis, necrosis,
necroptosis, autophagy, pyroptosis and ferroptosis (14–16,85,86).
A central focus of research is potential interactions between cell
death pathways and the identification of common upstream regulatory
mechanisms (101). Cells proceed
through different death pathways at distinct stages of IR. For
example, in cerebral ischemic stroke, brain tissue exhibits varying
patterns of cell death based on ischemic duration, oxygenation
levels and residual blood flow (96,97).
Moderate injury may activate autophagy as a protective mechanism to
preserve cell viability and facilitate recovery. By contrast,
severe injury typically leads to irreversible necrosis, apoptosis
and ferroptosis (102,103). ALDH2 is a key regulator of
cellular autophagy, apoptosis, necroptosis and ferroptosis during
IRI (7,20,37).
A growing body of evidence underscores the dynamic nature of cell
death processes in IRI and highlights the multifaceted role of
ALDH2 in regulating cell fate in various pathological contexts
(15,101–103). Further investigation into the
interplay between cell death pathways and ALDH2-mediated regulation
may unveil novel therapeutic strategies for mitigating IRI.
Apoptosis, a form of programmed cell death, is
characterized by morphological changes and is energy-dependent. In
the early stages, apoptosis is marked by cell shrinkage, chromatin
pyknosis (condensation), a reduction in cell volume and an increase
in cytoplasmic density, along with more compact organelles
(101,104). Pyknosis of chromatin, or the
condensation of nuclear material, is a hallmark of apoptosis.
Biochemically, apoptosis typically progresses through a
caspase-dependent cascade (101,104). Historically, apoptosis has been
considered to occur through two main pathways, the extrinsic (death
receptor-mediated) and the intrinsic (mitochondria-mediated)
pathway. Both pathways activate caspases, which initiate the
apoptotic process (101,105). The extrinsic pathway involves
interaction of cell surface death receptors [for example, Fas/CD95;
TNF-related apoptosis-inducing ligand-receptor (TRAIL-R);
TNF-receptor1 (TNFR1)] with their respective ligands, triggering
recruitment of proteins that activate caspases (101,105). By contrast, the intrinsic pathway
is driven by mitochondrial outer membrane permeabilization and loss
of mitochondrial integrity, a process regulated by the Bcl-2 family
of proteins. This pathway is characterized by balance between
pro-apoptotic proteins such as Bax and anti-apoptotic proteins such
as Bcl-2. Following mitochondrial permeabilization, pro-apoptotic
factors, including cytochrome c, are released into cytosol,
leading to the formation of a caspase-activating complex (101,105). These apoptotic pathways have been
implicated in IRI, as studies have shown their involvement in organ
damage during ischemia and reperfusion (106,107). ALDH2 is a key regulator of
apoptosis in IRI in different organs. In a myocardial IRI model,
ALDH2 activation was shown to reduce the production of toxic
aldehydes such as 4-HNE and MDA, protecting the myocardium via the
ERK/p38 signaling pathway (3). In
a renal IRI model, ALDH2 activation attenuates inflammation and
decreases kidney injury by modulating the IκBα/NF-κB/IL-17C pathway
(4). Similarly, in the liver,
ALDH2 activation by Alda-1 induces autophagy via the AKT/mTOR and
AMPK pathways, thereby protecting the liver from IRI-induced
hepatocyte apoptosis (11).
Furthermore, the role of ALDH2 in apoptosis regulation extends to
the nervous system (15). In the
intrinsic apoptotic pathway, the AKT signaling pathway regulates
balance between pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins
(108). In a pig heart IR model,
upregulation of ALDH2 increases Bcl-2/Bax ratio and decreases
activation of caspase-3 (109).
In humans, ALDH2 activation enhances mitochondrial membrane
potential via the PI3K/AKT/mTOR pathway, thereby decreasing ROS
production and inhibiting IR-induced apoptosis of cardiomyocytes
(18).
Cell death is typically classified into two
categories: RCD and accidental cell death (ACD). Apoptosis
represents RCD, while necrosis, driven by non-physiological stimuli
such as physical, mechanical or chemical stressors, typifies ACD
(110). Necrosis is characterized
by cell swelling, membrane rupture and release of cytoplasmic
contents. Necroptosis, a form of programmed necrosis, is primarily
regulated by post-translational modification of
receptor-interacting protein kinase (RIPK) 1, RIPK3 and mixed
lineage kinase domain-like protein (MLKL) (111). The TNF signaling pathway is key
for necroptosis. Following TNF binding to TNFR1, a cascade leads to
the formation of complex I, which includes TNF-receptor-associated
death domain protein, TNF-receptor-associated factor 2, cellular
inhibitor of apoptosis protein 1 or 2 and the linear ubiquitin
chain assembly complex. This complex facilitates linear
ubiquitination of receptor-interacting serine/threonine-protein
kinase 1 at the K63 site, enabling recruitment of downstream
proteins (112). If complex I
formation is disrupted, TNF signaling activates complex IIa or IIb,
which comprise Fas-associated protein with death domain and
caspase-8, proteins that have key roles in apoptosis execution
(113). Other stimuli, including
death receptors such as Fas [CD95 or apoptosis-related protein
(Apo)-1], death receptor 3 (DR3) (Apo-3), DR4 (Apo-2 or TRAIL-R1),
DR5 (TRAIL-R2) and DR6, initiate necroptosis via RIPK3.
Additionally, pattern recognition receptors, including toll-like
receptor (TLR) 3 and TLR4, mediate necroptosis through the
formation of necroptotic bodies via toll/interleukin-receptor
(TRIF), IFN-β, RIPK3 and MLKL, especially in the presence of
caspase inhibitors (113).
The inhibition of ROS clearance and calcium overload
triggers necroptosis through inflammatory factors such as TNF-α.
RIP3, a key factor in necroptosis, can also drive inflammation and
ROS production (114,115). Necroptosis generally occurs
during the late reperfusion phase and can persist for an extended
period (34). In the early
reperfusion phase (within 10 min), inhibiting RIP3 does not
mitigate cardiac injury, suggesting that during the initial phase
of IRI, RIP3 primarily mediates damage through the regulation of
oxidative stress and mitochondrial function, rather than
necroptosis (116). Studies have
revealed that the necroptosis inhibitor necrostatin-1 provides
notable protection in the late stages of infarction, improving IR
organ function over time, however, its efficacy is limited when
administered early during infarction (117,118).
ALDH2 inhibits necroptosis primarily by eliminating
superoxide and reactive aldehydes, thereby maintaining
intracellular Ca2+ homeostasis and stabilizing
mitochondrial function (119).
The protective effects of ALDH2 are mediated by several mechanisms:
ALDH2 downregulation activates caspase-associated pathways, which
subsequently promote necroptosis induction (120). Oxidative stress upregulates the
RIP1/RIP3/MLKL signaling cascade, leading to enhanced necroptosis.
In a glutamate-induced retinal excitotoxic model of glaucoma,
reduced ALDH2 expression is associated with increased expression of
RIP1/RIP3/MLKL signaling proteins, indicating elevated necroptosis
(121). In alcohol-induced
cardiac injury, decreased ALDH2 activity activates the caspase
pathway and necroptosis through RIP1/RIP3/MLKL signaling (122). Inhibition of ALDH2 activity leads
to the accumulation of 4-HNE, which promotes myocardial cell
necroptosis by inhibiting RIP1 ubiquitination and proteasomal
degradation. In mice, perfusion with 4-HNE exacerbates necroptosis
in a time- and concentration-dependent manner (123). Similarly, in high-glucose-induced
rats and cell necroptosis models, decreased ALDH2 expression is
associated with increased ROS production and upregulation of RIP1,
RIP3 and MLKL, resulting in enhanced necroptosis (7,124).
These findings highlight the key role of ALDH2 in mitigating
necroptosis via antioxidative and aldehyde detoxification
activities.
ALDH2 exhibits potent anti-inflammatory, anti-free
radical and anti-LPO activity, which intersect with ferroptosis
during IRI (39,98) (Fig.
5). ALDH2 serves a key role in mitigating ferroptosis by
metabolizing LPO products such as 4-HNE and MDA, thereby decreasing
oxidative stress-induced cellular damage. Additionally, ALDH2
enhances antioxidant defenses by activating SLC7A11, which promotes
intracellular GSH synthesis and decreases free iron
(Fe2+) levels, alleviating ferroptosis-mediated cell
injury (125). For example, in a
calcium oxalate kidney stone model, ALDH2 activation considerably
reduces renal crystal deposition and ferroptosis (125). In acute lung injury, ALDH2
provides protection by suppressing both pyroptosis and ferroptosis
(69). During post-cardiopulmonary
resuscitation organ damage, ALDH2 protects renal, intestinal and
pulmonary function by mitigating LPO and ferroptosis. Furthermore,
ALDH2 regulates the Nrf2/GPX4 signaling pathway to increase
cellular antioxidant capacity and diminish ferroptosis-associated
inflammatory damage. In periodontitis, ALDH2 activation alleviates
inflammation and promotes osteogenic differentiation of periodontal
ligament stem cells by inhibiting ferroptosis via Nrf2 activation
(126). Similarly, in gastric
ulcers, ALDH2 activation decreases inflammation and oxidative
stress while suppressing ferroptosis-associated damage via
inhibition of the nucleotide-binding domain, leucine-rich repeat
and pyrin domain-containing protein 3 inflammasome (127). Notably, ALDH2 deficiency or
decreased activity increases cellular sensitivity to ferroptosis
inducers or chemotherapeutic agents (128). In lung adenocarcinoma, decreased
ALDH2 expression increases sensitivity to platinum-based
chemotherapies by promoting ferroptosis (128).
Ferroptosis is driven by iron overload, free radical
generation, LPO and activation of cell death effectors, culminating
in membrane rupture. ALDH2 intervenes in multiple facets of ROS
clearance, inhibition of LPO and maintenance of mitochondrial
homeostasis during IR, all of which are associated with
ferroptosis. In IRI following cardiopulmonary resuscitation, ALDH2
effectively protects organ function by alleviating LPO and
ferroptosis in the kidney, intestine and lung (19,22).
ACSL4, a key enzyme in LPO associated with ferroptosis, is
regulated by ALDH2. In a murine model of Alzheimer's disease, ALDH2
suppresses special protein 1/ACSL4-mediated LPO and ferroptosis by
modulating downregulation of GPX4 and SLC7A11, while upregulating
NCOA4, thereby rescuing cardiac abnormality induced by amyloid
precursor protein/presenilin-1 mutations (129). During IR, LPO products such as
4-HNE and MDA serve as markers for ferroptosis. Specifically, 4-HNE
carbonylates cysteine residues at C93 of GPX4 and C247 of OTUD5,
hindering their interaction and aggravating K48-linked
polyubiquitin-related degradation of GPX4. ALDH2 activation
mitigates this effect, preserving GPX4 function and inhibiting
ferroptosis (35). ALDH2 also
stabilizes mitochondrial morphology and function, protecting
against cardiac dysfunction by activating the Nrf1-FUN14
domain-containing protein 1 (FUNDC1) cascade (130). Further mechanistic research
reveals that FUNDC1 interacts with GPX4 to facilitate its
recruitment into mitochondria via the translocase of the outer
membrane/translocase of the inner membrane (TIM) complex, enhancing
mitochondrial protection by ALDH2 (36). Regarding iron metabolism,
intracellular and extracellular ALDH2 decreases 4-HNE, activates
the Nrf2/HO-1 pathway to decrease intracellular iron accumulation
and mitigates hepatic cell ferroptosis through Parkin-mediated
mitochondrial autophagy (131).
Additionally, ALDH2 is associated with serum ferritin levels; ALDH2
mutant genotypes are associated with lower serum ferritin levels in
human males (132). These
findings underscore the multifaceted role of ALDH2 in modulating
ferroptosis and highlight its potential as a therapeutic target for
IRI.
Therapies targeting ALDH2 date from the late 1940s
with the introduction of disulfiram for the treatment of alcohol
addiction (30). Disulfiram
functions as a non-selective ALDH2 inhibitor, causing acetaldehyde
accumulation following alcohol consumption, which induces symptoms
such as nausea, vomiting and facial flushing (31). Despite early adoption of ALDH2
inhibitors such as disulfiram, to the best of our knowledge, no
clinically approved ALDH2 agonists exist for the treatment of IRI.
In IRI, ALDH2 expression is often decreased and its enzymatic
activity is impaired, leading to exacerbated tissue damage.
However, development of ALDH2 agonists offers promising potential
for the prevention and treatment of IRI. These agonists may restore
ALDH2 activity, mitigate oxidative stress and alleviate IR-induced
tissue injury (133–135). Research into ALDH2 agonists as
therapeutic agents for IRI is essential for identifying viable
treatment options in clinical practice. Determining the mechanisms
underlying ALDH2 activation and its protective effects against IRI
may facilitate development of novel compounds that target ALDH2
activation, paving the way for clinically applicable therapy.
The decreased activity of the ALDH2 2*2 variant
stems from a single amino acid substitution, where lysine replaces
glutamate, resulting in a 200-fold increase in the KM
(which represents the affinity of an enzyme for its substrate) for
NAD+ binding. Research has shown that Alda-1 does not
bind near the site of the ALDH2 2*2 mutation but rather closer to
the exit of the substrate-binding tunnel (135). Its binding site overlaps with
that of daidzin and extends toward the active site, leaving key
residues such as Cys302 and Glu268 unmodified, enabling them to
perform catalytic functions (135). The E487K substitution in ALDH2
2*2, where glutamate is replaced by lysine at position 487, leads
to decreased electron density near key residues, particularly those
involved in forming helix αG (residues 245–262) and the active-site
loop (residues 466–478). While Alda-1 does not interact directly
with these residues, it facilitates restoration of the α helix and
loop structures, bringing them closer to their native conformation
(135). By restricting substrate
diffusion within the substrate-to-coenzyme tunnel, Alda-1 increases
the effective concentration of reactive groups at the active site,
thereby enhancing dehydrogenase activity (135). This mechanism illustrates how
Alda-1 can restore enzymatic function in the ALDH2 2*2 variant,
presenting therapeutic potential for conditions associated with
ALDH2 dysfunction, such as IRI.
4-HNE serves as both a substrate for ALDH2 and an
inhibitor of its activity. ALDH2 contains a reactive Cys
nucleophile in its active site, which is modified by 4-HNE. This
lipid aldehyde forms covalent adducts by modifying protein thiols
and amines. At low micromolar concentrations, inhibition of ALDH2
by 4-HNE is reversible, but at higher concentrations, when the
active site thiols are covalently modified, inhibition becomes
irreversible (136). Alda-1 has
been shown to preserve ALDH2 catalytic activity in the presence of
high concentrations of 4-HNE, protecting the enzyme from
4-HNE-induced inactivation (26).
This protective effect is likely due to the ability of Alda-1 to
decrease interaction of 4-HNE with key residues such as Cys302,
Cys301 and Cys303, thereby preventing formation of aldehyde adducts
(26). This mechanism reveals how
Alda-1 maintains activity in structurally intact ALDH2,
distinguishing it from its effect on the ALDH2 2*2 variant.
GTN is used in clinical practice to treat
conditions such as angina pectoris, myocardial infarction and heart
failure (138). Its therapeutic
effects are primarily mediated by bioactivation of GTN in vascular
smooth muscle cells and platelets, leading to vasodilation of large
veins and arteries, inhibition of platelet aggregation and
increased levels of nitric oxide (NO) or S-nitrosothiol, as well as
elevated cyclic guanosine monophosphate levels (139). However, prolonged use of organic
nitrates such as GTN can result in development of nitrate
tolerance, characterized by increased oxidative stress, endothelial
dysfunction and sympathetic nerve activation (139,140). ALDH2 is the primary enzyme
responsible for bioactivating organic nitrates, catalyzing the
conversion of GTN to NO (141).
In human individuals with ALDH2-inactivating genotypes (ALDH2 1*2
and ALDH2 2*2) exhibit decreased bioactivation of GTN, which leads
to reduced vasodilatory effects (142). A clinical trial indicated that
ALDH2 gene polymorphisms affect the pharmacokinetics and
hemodynamics of GTN in human participants (143). In cardiovascular IRI, the
accumulation of free radicals exacerbates NO reactions with
superoxide, leading to the formation of peroxynitrite, which causes
nitration of ALDH2. This nitration impairs ALDH2 catalytic activity
and contributes to nitrate tolerance (144). Nitrates, including GTN, can
inhibit ALDH2 activity (139,140). In vitro, GTN rapidly and
effectively inactivates ALDH2 (145). Additionally, in vivo a
study in nitrate-tolerant rats revealed an association between
nitrate tolerance, ROS accumulation and loss of ALDH2 activity
(146). Therefore, when targeting
ALDH2 therapeutically, it is important to consider potential
antagonistic or competitive interactions with clinically used
drugs, such as nitrates. Strategies aimed at modulating ALDH2
activity should be evaluated to avoid unintended consequences, as
interventions that appear beneficial in one context may have
adverse effects in another.
ISO is a commonly used volatile anesthetic with a
well-established safety profile that exerts protective effects in
IRI across various types of organs. ISO therapy can mitigate
infarction volume and intracranial hemorrhage in tissue-type
plasminogen activator-exaggerated brain injury at low
concentrations (147). ISO may be
associated with a lower rate of postoperative myocardial infarction
and hemodynamic complications in patients undergoing coronary
artery bypass grafting (148).
Protective effects of ISO have also been observed in the heart
(149), kidney (150) and liver (151). The mechanism underlying
protective effects of ISO in IRI may involve the activation of
ALDH2. ISO pretreatment decreases mitochondrial translocation of
PKCδ and enhances ALDH2 phosphorylation (152). This leads to decreased release of
myocardial injury markers such as lactate dehydrogenase and
creatine kinase-myocardial band. Conversely, activation of PKCε
facilitates the protective effects of ISO (152). These findings suggest that ISO
exerts its protective effects in IRI through ALDH2 activation and
modulation of PKC signaling pathways, offering insight into
potential therapeutic strategies for mitigating tissue damage in
IRI.
IPC refers to the phenomenon where brief episodes
of ischemia before prolonged ischemic events confer cell protection
against subsequent IRI (39). To
the best of our knowledge, there are no effective clinical
interventions for established IRI; however, IPC has been studied as
a potential therapeutic strategy to mitigate tissue damage
associated with IRI (140–145).
In 1997, a clinical trial showed that IPC exerts functional
protection against simulated IRI (153); numerous trials have confirmed its
effectiveness in alleviating IRI in the heart, kidney, intestine
and liver (154–156). Genetic or pharmacological
inhibition of ALDH2 impairs the cardioprotective effects of IPC,
highlighting the role of ALDH2 activation in mediating protection
against IRI (157). The
protective mechanism of ALDH2 in IPC involves sequential activation
of PKCε and ALDH2 (158). PKCε
translocates into the mitochondria, where it interacts with ALDH2,
increasing its enzyme activity (159). This interaction is essential for
the protective effects of IPC against IRI. Overall, these findings
underscore the central role of ALDH2 activation in IPC-mediated
protection against IRI and offer insights into the mechanisms
involved. Further research into ALDH2 activation may facilitate
development of novel therapeutic strategies to prevent and treat
IRI.
Ethanol and acetaldehyde exposure are
pharmacological interventions that mimic the cardioprotective
effects of IPC, although the effects are limited at lower doses.
For example, brief exposure to ethanol (10–50 mM) before ischemia
has been shown to reduce myocardial damage caused by prolonged
ischemia (160). Cardioprotective
effects of ethanol are mediated by activation of ALDH2 (161). Similarly, pretreatment with 50 µM
acetaldehyde enhances ALDH2 activity and decreases myocardial
damage in wild-type mouse hearts, without considerably altering
cardiac acetaldehyde levels (157). However, in ALDH2 2*2 mice,
acetaldehyde pretreatment leads to a three-fold increase in cardiac
acetaldehyde levels, exacerbating IRI (157). The underlying mechanism of ALDH2
activation by ethanol and acetaldehyde involves activation of PKCε
in cardiomyocytes (157,160).
In addition to ethanol and acetaldehyde, other
compounds activate ALDH2, although the exact mechanisms remain
incompletely understood. Estrogen (162), heat shock factor 1 (163) and melatonin (164) have been proposed to increase
ALDH2 activity by promoting mitochondrial import of PKCε.
Antioxidants such as lipoic acid (130), resveratrol (165) and vitamins D (166), C (167) and E (168) activate ALDH2 through various
signaling pathways. Notably, flurbiprofen, a non-steroidal
anti-inflammatory drug, activates ALDH2 in the context of its
anti-obesity effects (169). In
summary, pharmacological activation of ALDH2 offers potential
therapeutic avenues for IRI; further investigation into specific
drugs targeting ALDH2 activation is essential for minimizing side
effects and improving clinical outcomes.
ALDH activity is notably elevated in certain stem
or progenitor cells, designated ALDHbr cells. These cells have been
identified and isolated from numerous types of normal tissue,
including human cord blood, bone marrow, mobilized peripheral
blood, skeletal muscle and breast tissue, as well as rodent brain,
pancreas and prostate tissue, using flow cytometry techniques
(141,170). In a preclinical ischemic disease
model ALDHbr cell administration has considerable efficacy in
promoting tissue repair and local neovascularization and may have
clinical application (170). In
patients with ischemic heart failure, ALDHbr cell therapy is
considered safe, potentially enhancing perfusion and functional
recovery (171). Additionally,
clinical trials investigating intermittent claudication have
reported therapeutic effects consistent with ALDH2 isozyme
functions, such as improved ischemic tolerance, angiogenesis and
mitochondrial metabolism (172,173). However, the specific ALDH isozyme
targeted by these therapies is unclear. PCR array has demonstrated
a marked increase in ALDH2 expression in ALDHbr cells following
ischemic injury (174). By
contrast, a clinical trial involving patients with claudication and
infrainguinal peripheral artery disease, who received ten
injections of ALDHbr in the thigh and calf, showed no improvement
in peripheral artery disease outcomes, potentially due to sample
size limitations (173). While
ALDHbr cell therapy offers promising therapeutic potential, its
widespread application is constrained by its high cost and
complexity (173), with modest
effects on clinical outcome.
Targeting ferroptosis is a promising strategy for
mitigating IRI. Several small-molecule compounds designed to
intervene at various stages of the ferroptotic cascade have been
developed. Deferoxamine, a widely studied iron chelator, has
demonstrated considerable efficacy in inhibiting ferroptosis
(175–177). Deferoxamine decreases lactate
dehydrogenase release, highlighting its therapeutic potential in
alleviating tissue damage caused by IR in isolated wild-type mouse
hearts (175). In a randomized
controlled trial, deferoxamine effectively limited generation of
ROS by chelating redox−active iron, thereby alleviating
oxidative stress without reducing infarct size (176). Dexrazoxane, the only US Food and
Drug Administration-approved drug for doxorubicin-induced
cardiotoxicity, also serves as an iron chelator. In a murine model
of doxorubicin-induced cardiomyopathy and IR, dexrazoxane decreases
non-heme iron levels in the heart and mitigates doxorubicin-induced
liver iron accumulation (177).
Ferrostatin-1 (Fer-1) has been identified as a potent inhibitor of
LPO (178). Li et al
(178) reported that Fer-1
decreases levels of pro-ferroptotic AA-PE, thereby preventing
cardiomyocyte death. Additionally, by inhibiting the TLR4/TRIF/type
I IFN signaling pathway associated with ferroptosis, Fer-1 impedes
neutrophil adhesion to coronary endothelial cells, thereby
mitigating inflammation (178).
Liproxstatin-1 is another selective ferroptosis inhibitor.
Administering Liproxstatin-1 1 h before ischemia in a mouse model
of intestinal IRI induces expression of GPX4 and decreases COX2
expression, as well as levels of 12- and 15-hydroxyeicosatetraenoic
acid, LPO products and serum markers such as lactate dehydrogenase,
TNF-α and IL-6 (179). Several
drugs target multiple stages of ferroptosis simultaneously. For
example, puerarin, used clinically for heart failure, exerts a
multifaceted protective effect against IRI. Liu et al
(180) showed that puerarin
decreases erastin-induced ferroptosis in cardiomyocytes by
downregulating NADPH oxidase 4 expression, upregulating GPX4 and
inhibiting both iron overload and LPO. In Alzheimer's disease, the
beneficial effects of puerarin are associated with changes in
expression of iron metabolism-associated proteins in neuronal
cells, including divalent metal ion transporter 1 and TFR1. By
mitigating iron metabolism-related damage, puerarin improves
cognitive function and memory in neurodegenerative disease
(181). Despite these advances,
no specific drug targeting ferroptosis has been developed for
treatment of IRI. Further research into the specific regulatory
mechanisms and targets of ferroptosis in IRI is essential for
identifying potential therapeutic strategies. Additionally,
upstream signaling pathways that regulate ferroptosis remain
unclear and the interaction between ALDH2 and ferroptosis, along
with the precise mechanisms involved, have yet to be fully
elucidated. Given the dynamic nature of pathological changes in
IRI, temporal progression of ferroptosis during IRI should be
further investigated. Potential therapies targeting ferroptosis are
summarized in Table II.
IRI is a complex complication in clinical practice,
where damage initiated during ischemia can persist and worsen upon
reperfusion. The mechanisms underlying IRI involve a number of
cellular injury processes, including autophagy, necrosis,
apoptosis, necroptosis and ferroptosis (101). Among the key factors in this
cascade, ALDH2 is a cellular oxidoreductase responsible for
acetaldehyde metabolism, which is associated with antioxidant
defense (28). The present review
highlights the key role of ALDH2 in IRI, particularly in scavenging
ROS and limiting LPO accumulation. Additionally, ALDH2 regulates
cell death pathways under oxidative stress conditions (7,20).
The interaction between ALDH2 and ferroptosis suggests these as
potential therapeutic targets for mitigating IRI. The present
review summarizes the role of ALDH2 in regulation of ferroptosis
and other cell death pathways, aiming to uncover intersections with
ferroptosis and enhance understanding of its role in IRI. However,
the mechanisms by which activators of ALDH2 and ferroptosis
inhibitors mitigate IRI remain incompletely understood. To the best
of our knowledge, clinical studies on the role of ALDH2 and
ferroptosis in IRI are scarce. Moreover, the aforementioned studies
research on these compounds has primarily been conducted in rodent
models of IR, highlighting the need for clinical trials to evaluate
their safety and efficacy. Future research should investigate the
detailed mechanisms through which ALDH2 and ferroptosis inhibitors
protect against IRI to develop more effective treatment strategies
and improve management and prevention of IRI in clinical
practice.
The authors would like to thank Dr Foquan Luo and
Dr Xiaomin Wu [Center for Rehabilitation Medicine, Department of
Anesthesiology, Zhejiang Provincial People's Hospital (Affiliated
People's Hospital, Hangzhou Medical College)] for their assistance
in data collection.
Funding: No funding was received.
Not applicable.
LH constructed figures and wrote the manuscript. WZ
conceived and designed the study and edited the manuscript. Data
authentication is not applicable. Both authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Panisello-Rosello A, Lopez A, Folch-Puy E,
Carbonell T, Rolo A, Palmeira C, Adam R, Net M and Roselló-Catafau
J: Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury:
An update. World J Gastroenterol. 24:2984–2994. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang M, Liu Q, Meng H, Duan H, Liu X, Wu
J, Gao F, Wang S, Tan R and Yuan J: Ischemia-reperfusion injury:
Molecular mechanisms and therapeutic targets. Signal Transduct
Target Ther. 9:122024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bai X, Zhang J, Yang H, Linghu K and Xu M:
SNHG3/miR-330-5p/HSD11B1 alleviates myocardial ischemia-reperfusion
injury by regulating the ERK/p38 signaling pathway. Protein Pept
Lett. 30:699–708. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Xiong Y, Luo J, Hu Q, Lan J, Zou
Y, Ma Q, Yao H, Liu Z, Zhong Z, et al: Aldehyde dehydrogenase 2
protects the kidney from Ischemia-reperfusion injury by suppressing
the I κ B α/NF-κ B/IL-17C pathway. Oxid Med Cell Longev.
2023:22640302023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Diao M, Xu J, Wang J, Zhang M, Wu C, Hu X,
Zhu Y, Zhang M and Hu W: Alda-1, an activator of ALDH2, improves
postresuscitation cardiac and neurological outcomes by inhibiting
pyroptosis in swine. Neurochem Res. 47:1097–1099. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao R, Lv C, Qu Y, Yang H, Hao C, Sun X,
Hu X, Yang Y and Tang Y: Remote ischemic conditioning mediates
cardio-protection after myocardial ischemia/reperfusion injury by
reducing 4-HNE levels and regulating autophagy via the
ALDH2/SIRT3/HIF1alpha signaling pathway. J Cardiovasc Transl Res.
17:169–182. 2024.PubMed/NCBI
|
|
7
|
Kang P, Wang J, Fang D, Fang T, Yu Y,
Zhang W, Shen L, Li Z, Wang H, Ye H and Gao Q: Activation of ALDH2
attenuates high glucose induced rat cardiomyocyte fibrosis and
necroptosis. Free Radic Biol Med. 146:198–210. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li M, Xu M, Li J, Chen L, Xu D, Tong Y,
Zhang J, Wu H, Kong X and Xia Q: Alda-1 ameliorates liver
Ischemia-Reperfusion injury by activating aldehyde dehydrogenase 2
and enhancing autophagy in mice. J Immunol Res. 2018:98071392018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin D, Xiang T, Qiu Q, Leung J, Xu J, Zhou
W, Hu Q, Lan J, Liu Z, Zhong Z, et al: Aldehyde dehydrogenase 2
regulates autophagy via the Akt-mTOR pathway to mitigate renal
ischemia-reperfusion injury in hypothermic machine perfusion. Life
Sci. 253:1177052020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin L, Tao JP, Li M, Peng J, Zhou C,
Ouyang J and Si YY: Mechanism of ALDH2 improves the neuronal damage
caused by hypoxia/reoxygenation. Eur Rev Med Pharmacol Sci.
26:2712–2720. 2022.PubMed/NCBI
|
|
11
|
Liu Z, Ye S, Zhong X, Wang W, Lai CH, Yang
W, Yue P, Luo J, Huang X, Zhong Z, et al: Pretreatment with the
ALDH2 activator Alda-1 protects rat livers from
ischemia/reperfusion injury by inducing autophagy. Mol Med Rep.
22:2373–2385. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma LL, Ding ZW, Yin PP, Wu J, Hu K, Sun
AJ, Zou YZ and Ge JB: Hypertrophic preconditioning cardioprotection
after myocardial ischaemia/reperfusion injury involves
ALDH2-dependent metabolism modulation. Redox Biol. 43:1019602021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan G, Roy B and Palaniyandi SS: Diabetic
aldehyde dehydrogenase 2 Mutant (ALDH2*2) mice are more susceptible
to cardiac Ischemic-Reperfusion injury due to 4-Hydroxy-2-Nonenal
induced coronary endothelial cell damage. J Am Heart Assoc.
10:e0211402021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Papatheodorou I, Galatou E, Panagiotidis
GD, Ravingerova T and Lazou A: Cardioprotective effects of
PPARbeta/delta activation against ischemia/reperfusion injury in
rat heart are associated with ALDH2 upregulation, amelioration of
oxidative stress and preservation of mitochondrial energy
production. Int J Mol Sci. 22:e0211402021. View Article : Google Scholar
|
|
15
|
Qu Y, Liu Y and Zhang H: ALDH2 activation
attenuates oxygen-glucose deprivation/reoxygenation-induced cell
apoptosis, pyroptosis, ferroptosis and autophagy. Clin Transl
Oncol. 25:3203–1326. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sidramagowda Patil S, Hernandez-Cuervo H,
Fukumoto J, Krishnamurthy S, Lin M, Alleyn M, Breitzig M, Narala
VR, Soundararajan R, Lockey RF, et al: Alda-1 attenuates
hyperoxia-induced acute lung injury in mice. Front Pharmacol.
11:5979422020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun X, Gao R, Li W, Zhao Y, Yang H, Chen
H, Jiang H, Dong Z, Hu J, Liu J, et al: Alda-1 treatment promotes
the therapeutic effect of mitochondrial transplantation for
myocardial ischemia-reperfusion injury. Bioact Mater. 6:2058–2069.
2021.PubMed/NCBI
|
|
18
|
Tan X, Chen YF, Zou SY, Wang WJ, Zhang NN,
Sun ZY, Xian W, Li XR, Tang B, Wang HJ, et al: ALDH2 attenuates
ischemia and reperfusion injury through regulation of mitochondrial
fusion and fission by PI3K/AKT/mTOR pathway in diabetic
cardiomyopathy. Free Radic Biol Med. 195:219–230. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu H, Xu S, Diao M, Wang J, Zhang G and Xu
J: Alda-1 treatment alleviates lung injury after cardiac arrest and
resuscitation in swine. Shock. 58:464–469. 2022.PubMed/NCBI
|
|
20
|
Xu T, Guo J, Wei M, Wang J, Yang K, Pan C,
Pang J, Xue L, Yuan Q, Xue M, et al: Aldehyde dehydrogenase 2
protects against acute kidney injury by regulating autophagy via
the Beclin-1 pathway. JCI Insight. 6:e1381832021.PubMed/NCBI
|
|
21
|
Yoval-Sanchez B, Calleja LF, de la Luz
Hernandez-Esquivel M and Rodriguez-Zavala JS: Piperlonguminine a
new mitochondrial aldehyde dehydrogenase activator protects the
heart from ischemia/reperfusion injury. Biochim Biophys Acta Gen
Subj. 1864:1296842020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Q, Gao J, Shao X, Lu W, Chen L and Jin
L: The Effects of Alda-1 treatment on renal and intestinal injuries
after cardiopulmonary resuscitation in pigs. Front Med (Lausanne).
9:8924722022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang R, Xue MY, Liu BS, Wang WJ, Fan XH,
Zheng BY, Yuan QH, Xu F, Wang JL and Chen YG: Aldehyde
dehydrogenase 2 preserves mitochondrial morphology and attenuates
hypoxia/reoxygenation-induced cardiomyocyte injury. World J Emerg
Med. 11:246–254. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang ZX, Li H, He JS, Chu HJ, Zhang XT
and Yin L: Remote ischemic postconditioning alleviates myocardial
ischemia/reperfusion injury by up-regulating ALDH2. Eur Rev Med
Pharmacol Sci. 22:6475–6484. 2018.PubMed/NCBI
|
|
25
|
Zhou T, Wang X, Wang K, Lin Y, Meng Z, Lan
Q, Jiang Z, Chen J, Lin Y, Liu X, et al: Activation of aldehyde
dehydrogenase-2 improves ischemic random skin flap survival in
rats. Front Immunol. 14:11276102023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CH, Ferreira JC, Gross ER and
Mochly-Rosen D: Targeting aldehyde dehydrogenase 2: New therapeutic
opportunities. Physiol Rev. 94:1–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lamb RJ, Griffiths K, Lip GYH, Sorokin V,
Frenneaux MP, Feelisch M and Madhani M: ALDH2 polymorphism and
myocardial infarction: From alcohol metabolism to redox regulation.
Pharmacol Ther. 259:1086662024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoshida A, Hsu LC and Yasunami M: Genetics
of human alcohol-metabolizing enzymes. Prog Nucleic Acid Res Mol
Biol. 40:255–287. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mali VR and Palaniyandi SS: Regulation and
therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart
disease. Free Radic Res. 48:251–263. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schneider C, Porter NA and Brash AR:
Routes to 4-hydroxynonenal: Fundamental issues in the mechanisms of
lipid peroxidation. J Biol Chem. 283:15539–15543. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kimura M, Yokoyama A and Higuchi S:
Aldehyde dehydrogenase-2 as a therapeutic target. Expert Opin Ther
Targets. 23:955–966. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ke K, Li L, Lu C, Zhu Q, Wang Y, Mou Y,
Wang H and Jin W: The crosstalk effect between ferrous and other
ions metabolism in ferroptosis for therapy of cancer. Front Oncol.
12:9160822022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang Q, Yi X, Zhu XH, Wei X and Jiang DS:
Regulated cell death in myocardial ischemia-reperfusion injury.
Trends Endocrinol Metab. 35:219–234. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu L, Pang J, Qin D, Li R, Zou D, Chi K,
Wu W, Rui H, Yu H, Zhu W, et al: Deubiquitinase OTUD5 as a novel
protector against 4-HNE-triggered ferroptosis in myocardial
ischemia/reperfusion injury. Adv Sci (Weinh). 10:e23018522023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bi Y, Liu S, Qin X, Abudureyimu M, Wang L,
Zou R, Ajoolabady A, Zhang W, Peng H, Ren J and Zhang Y: FUNDC1
interacts with GPx4 to govern hepatic ferroptosis and fibrotic
injury through a mitophagy-dependent manner. J Adv Res. 55:45–60.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu W, Feng D, Shi X, Wei Q and Yang L:
The potential role of mitochondrial acetaldehyde dehydrogenase 2 in
urological cancers from the perspective of ferroptosis and cellular
senescence. Front Cell Dev Biol. 10:8501452022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Droge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen CH, Sun L and Mochly-Rosen D:
Mitochondrial aldehyde dehydrogenase and cardiac diseases.
Cardiovasc Res. 88:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wilson DF and Matschinsky FM: Ethanol
metabolism: The good, the bad, and the ugly. Med Hypotheses.
140:1096382020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hink U, Daiber A, Kayhan N, Trischler J,
Kraatz C, Oelze M, Mollnau H, Wenzel P, Vahl CF, Ho KK, et al:
Oxidative inhibition of the mitochondrial aldehyde dehydrogenase
promotes nitroglycerin tolerance in human blood vessels. J Am Coll
Cardiol. 50:2226–2232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu Y, Yuan Q, Cao S, Cui S, Xue L, Song X,
Li Z, Xu R, Yuan Q and Li R: Aldehyde dehydrogenase 2 inhibited
oxidized LDL-induced NLRP3 inflammasome priming and activation via
attenuating oxidative stress. Biochem Biophys Res Commun.
529:998–1004. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Moldovan L and Moldovan NI: Oxygen free
radicals and redox biology of organelles. Histochem Cell Biol.
122:395–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bielski BH, Arudi RL and Sutherland MW: A
study of the reactivity of HO2/O2-with unsaturated fatty acids. J
Biol Chem. 258:4759–4761. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Browne RW and Armstrong D: HPLC analysis
of lipid-derived polyunsaturated fatty acid peroxidation products
in oxidatively modified human plasma. Clin Chem. 46:829–836. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Schneider C, Boeglin WE, Yin H, Porter NA
and Brash AR: Intermolecular peroxyl radical reactions during
autoxidation of hydroxy and hydroperoxy arachidonic acids generate
a novel series of epoxidized products. Chem Res Toxicol.
21:895–903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Singh S, Brocker C, Koppaka V, Chen Y,
Jackson BC, Matsumoto A, Thompson DC and Vasiliou V: Aldehyde
dehydrogenases in cellular responses to oxidative/electrophilic
stress. Free Radic Biol Med. 56:89–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gao J, Hao Y, Piao X and Gu X: Aldehyde
dehydrogenase 2 as a therapeutic target in oxidative stress-related
diseases: Post-translational modifications deserve more attention.
Int J Mol Sci. 23:26822022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Breitzig M, Bhimineni C, Lockey R and
Kolliputi N: 4-Hydroxy-2-nonenal: A critical target in oxidative
stress? Am J Physiol Cell Physiol. 311:C537–C543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schaur RJ, Siems W, Bresgen N and Eckl PM:
4-Hydroxy-nonenal-a bioactive lipid peroxidation product.
Biomolecules. 5:2247–337. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Forman HJ: Reactive oxygen species and
alpha, beta-unsaturated aldehydes as second messengers in signal
transduction. Ann N Y Acad Sci. 1203:35–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shoeb M, Ansari NH, Srivastava SK and
Ramana KV: 4-Hydroxynonenal in the pathogenesis and progression of
human diseases. Curr Med Chem. 21:230–237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang H and Forman HJ: Signaling pathways
involved in phase II gene induction by alpha, beta-unsaturated
aldehydes. Toxicol Ind Health. 25:269–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dalleau S, Baradat M, Gueraud F and Huc L:
Cell death and diseases related to oxidative stress:
4-hydroxynonenal (HNE) in the balance. Cell Death Differ.
20:1615–1630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xue Z, Zhao K, Sun Z, Wu C, Yu B, Kong D
and Xu B: Isorhapontigenin ameliorates cerebral
ischemia/reperfusion injury via modulating Kinase
Cepsilon/Nrf2/HO-1 signaling pathway. Brain Behav. 11:e021432021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang Y, Li B, Liu G, Han Q, Diao Y and Liu
J: Corilagin attenuates intestinal ischemia/reperfusion injury in
mice by inhibiting ferritinophagy-mediated ferroptosis through
disrupting NCOA4-ferritin interaction. Life Sci. 334:1221762023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Endo J, Sano M, Katayama T, Hishiki T,
Shinmura K, Morizane S, Matsuhashi T, Katsumata Y, Zhang Y, Ito H,
et al: Metabolic remodeling induced by mitochondrial aldehyde
stress stimulates tolerance to oxidative stress in the heart. Circ
Res. 105:1118–1127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Esterbauer H, Schaur RJ and Zollner H:
Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and
related aldehydes. Free Radic Biol Med. 11:81–128. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Giera M, Lingeman H and Niessen WM: Recent
advancements in the LC- and GC-Based analysis of malondialdehyde
(MDA): A brief overview. Chromatographia. 75:433–440. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Esterbauer H and Zollner H: Methods for
determination of aldehydic lipid peroxidation products. Free Radic
Biol Med. 7:197–203. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shen Z, Zhang Y, Bu G and Fang L: Renal
denervation improves mitochondrial oxidative stress and cardiac
hypertrophy through inactivating SP1/BACH1-PACS2 signaling. Int
Immunopharmacol. 141:1127782024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tsikas D: Assessment of lipid peroxidation
by measuring malondialdehyde (MDA) and relatives in biological
samples: Analytical and biological challenges. Anal Biochem.
524:13–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lankin VZ, Tikhaze AK and Melkumyants AM:
Malondialdehyde as an important key factor of molecular mechanisms
of vascular wall damage under heart diseases development. Int J Mol
Sci. 24:1282022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Busch CJ and Binder CJ: Malondialdehyde
epitopes as mediators of sterile inflammation. Biochim Biophys Acta
Mol Cell Biol Lipids. 1862:398–406. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Agadjanyan ZS, Dmitriev LF and Dugin SF: A
new role of phosphoglucose isomerase. Involvement of the glycolytic
enzyme in aldehyde metabolism. Biochemistry (Mosc). 70:1251–1255.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu H, Wu X, Luo J, Wang X, Guo H, Feng D,
Zhao L, Bai H, Song M, Liu X, et al: Pterostilbene attenuates
astrocytic inflammation and neuronal oxidative injury after
Ischemia-reperfusion by inhibiting NF-κB phosphorylation. Front
Immunol. 10:24082019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qi D, Chen P, Bao H, Zhang L, Sun K, Song
S and Li T: Dimethyl fumarate protects against hepatic
ischemia-reperfusion injury by alleviating ferroptosis via the
NRF2/SLC7A11/HO-1 axis. Cell Cycle. 22:818–828. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang IC, Lin JH, Lee WS, Liu CH, Lin TY
and Yang KT: Baicalein and luteolin inhibit
ischemia/reperfusion-induced ferroptosis in rat cardiomyocytes. Int
J Cardiol. 375:74–86. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cao Z, Qin H, Huang Y, Zhao Y, Chen Z, Hu
J and Gao Q: Crosstalk of pyroptosis, ferroptosis, and
mitochondrial aldehyde dehydrogenase 2-related mechanisms in
sepsis-induced lung injury in a mouse model. Bioengineered.
13:4810–4820. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yoval-Sanchez B and Rodriguez-Zavala JS:
Differences in susceptibility to inactivation of human aldehyde
dehydrogenases by lipid peroxidation byproducts. Chem Res Toxicol.
25:722–729. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang T, Zhao Q, Ye F, Huang CY, Chen WM
and Huang WQ: Alda-1, an ALDH2 activator, protects against hepatic
ischemia/reperfusion injury in rats via inhibition of oxidative
stress. Free Radic Res. 52:629–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ,
Wang G, Kurihara H, He RR, Li YF, Chen Y, et al: ALOX15-launched
PUFA-phospholipids peroxidation increases the susceptibility of
ferroptosis in ischemia-induced myocardial damage. Signal Transduct
Target Ther. 7:2882022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yan HF, Tuo QZ, Yin QZ and Lei P: The
pathological role of ferroptosis in ischemia/reperfusion-related
injury. Zool Res. 41:220–230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wu X, Li Y, Zhang S and Zhou X:
Ferroptosis as a novel therapeutic target for cardiovascular
disease. Theranostics. 11:3052–3059. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liu J, Kang R and Tang D: Signaling
pathways and defense mechanisms of ferroptosis. FEBS J.
289:7038–7050. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu C, Li Z, Li B, Liu W, Zhang S, Qiu K
and Zhu W: Relationship between ferroptosis and mitophagy in
cardiac ischemia reperfusion injury: A mini-review. PeerJ.
11:e149522023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liang C, Zhang X, Yang M and Dong X:
Recent progress in ferroptosis inducers for cancer therapy. Adv
Mater. 31:e19041972019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E49675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Haeggstrom JZ and Funk CD: Lipoxygenase
and leukotriene pathways: Biochemistry, biology, and roles in
disease. Chem Rev. 111:5866–5898. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Braughler JM, Duncan LA and Chase RL: The
involvement of iron in lipid peroxidation. Importance of ferric to
ferrous ratios in initiation. J Biol Chem. 261:10282–10289. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Matsuyama M, Nakatani T, Hase T, Kawahito
Y, Sano H, Kawamura M and Yoshimura R: The expression of
cyclooxygenases and lipoxygenases in renal ischemia-reperfusion
injury. Transplant Proc. 36:1939–1942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Jiang M, Jike Y, Liu K, Gan F, Zhang K,
Xie M, Zhang J, Chen C, Zou X, Jiang X, et al: Exosome-mediated
miR-144-3p promotes ferroptosis to inhibit osteosarcoma
proliferation, migration, and invasion through regulating ZEB1. Mol
Cancer. 22:1132023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yang R, Gao W, Wang Z, Jian H, Peng L, Yu
X, Xue P, Peng W, Li K and Zeng P: Polyphyllin I induced
ferroptosis to suppress the progression of hepatocellular carcinoma
through activation of the mitochondrial dysfunction via
Nrf2/HO-1/GPX4 axis. Phytomedicine. 122:1551352024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen X, Huang J, Yu C, Liu J, Gao W, Li J,
Song X, Zhou Z, Li C, Xie Y, et al: A noncanonical function of
EIF4E limits ALDH1B1 activity and increases susceptibility to
ferroptosis. Nat Commun. 13:63182022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Jang EJ, Jeong HO, Park D, Kim DH, Choi
YJ, Chung KW, Park MH, Yu BP and Chung HY: Src Tyrosine kinase
activation by 4-hydroxynonenal upregulates p38, ERK/AP-1 signaling
and COX-2 expression in YPEN-1 Cells. PLoS One. 10:e01292442015.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lee J and Hyun DH: The interplay between
intracellular iron homeostasis and neuroinflammation in
neurodegenerative diseases. Antioxidants (Basel). 12:9182023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Miyamoto HD, Ikeda M, Ide T, Tadokoro T,
Furusawa S, Abe K, Ishimaru K, Enzan N, Sada M, Yamamoto T, et al:
Iron overload via heme degradation in the endoplasmic reticulum
triggers ferroptosis in myocardial ischemia-reperfusion injury.
JACC Basic Transl Sci. 7:800–819. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ito J, Omiya S, Rusu MC, Ueda H, Murakawa
T, Tanada Y, Abe H, Nakahara K, Asahi M, Taneike M, et al: Iron
derived from autophagy-mediated ferritin degradation induces
cardiomyocyte death and heart failure in mice. Elife.
10:e621742021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Radic Biol Med.
162:339–352. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Masaldan S, Clatworthy SAS, Gamell C,
Meggyesy PM, Rigopoulos AT, Haupt S, Haupt Y, Denoyer D, Adlard PA,
Bush AI and Cater MA: Iron accumulation in senescent cells is
coupled with impaired ferritinophagy and inhibition of ferroptosis.
Redox Biol. 14:100–115. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li W, Li W, Wang Y, Leng Y and Xia Z:
Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated
ferritinophagy during diabetes myocardial ischemia/reperfusion
injury. Cell Death Discov. 7:2672021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Fan Z, Cai L, Wang S, Wang J and Chen B:
Baicalin prevents myocardial ischemia/reperfusion injury through
inhibiting ACSL4 mediated ferroptosis. Front Pharmacol.
12:6289882021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wu Y, Jiao H, Yue Y, He K, Jin Y, Zhang J,
Zhang J, Wei Y, Luo H, Hao Z, et al: Ubiquitin ligase E3 HUWE1/MULE
targets transferrin receptor for degradation and suppresses
ferroptosis in acute liver injury. Cell Death Differ. 29:1705–1718.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bresgen N, Jaksch H, Lacher H,
Ohlenschlager I, Uchida K and Eckl PM: Iron-mediated oxidative
stress plays an essential role in ferritin-induced cell death. Free
Radic Biol Med. 48:1347–1357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ma S, Sun L, Wu W, Wu J, Sun Z and Ren J:
USP22 Protects against myocardial Ischemia-Reperfusion Injury via
the SIRT1-p53/SLC7A11-dependent inhibition of Ferroptosis-induced
cardiomyocyte death. Front Physiol. 11:5513182020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Alim I, Caulfield JT, Chen Y, Swarup V,
Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie
EJ, et al: Selenium drives a transcriptional adaptive program to
block ferroptosis and treat stroke. Cell. 177:1262–1279.e25. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J,
Wang G, Guo Z, Ye Y and Guo F: Chondrocyte ferroptosis contribute
to the progression of osteoarthritis. J Orthop Translat. 27:33–43.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang Q, Jia M, Wang Y, Wang Q and Wu J:
Cell death mechanisms in cerebral Ischemia-Reperfusion injury.
Neurochem Res. 47:3525–3542. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Guo J, Tuo QZ and Lei P: Iron,
ferroptosis, and ischemic stroke. J Neurochem. 165:487–520. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:a0060802015.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pang Q, Zhao Y, Chen X, Zhao K, Zhai Q and
Tu F: Apigenin protects the brain against Ischemia/Reperfusion
injury via Caveolin-1/VEGF in vitro and in vivo. Oxid Med Cell
Longev. 2018:70172042018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Liu J, Li J, Yang Y, Wang X, Zhang Z and
Zhang L: Neuronal apoptosis in cerebral ischemia/reperfusion area
following electrical stimulation of fastigial nucleus. Neural Regen
Res. 9:727–734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Teijido O and Dejean L: Upregulation of
Bcl2 inhibits apoptosis-driven BAX insertion but favors BAX
relocalization in mitochondria. FEBS Lett. 584:3305–3310. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Andreka G, Vertesaljai M, Szantho G, Font
G, Piroth Z, Fontos G, Juhasz ED, Szekely L, Szelid Z, Turner MS,
et al: Remote ischaemic postconditioning protects the heart during
acute myocardial infarction in pigs. Hear. 93:749–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Guerrero-Mauvecin J, Villar-Gomez N,
Rayego-Mateos S, Ramos AM, Ruiz-Ortega M, Ortiz A and Sanz AB:
Regulated necrosis role in inflammation and repair in acute kidney
injury. Front Immunol. 14:13249962023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Annibaldi A and Meier P: Checkpoints in
TNF-Induced cell death: Implications in inflammation and cancer.
Trends Mol Med. 24:49–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Seo J, Nam YW, Kim S, Oh DB and Song J:
Necroptosis molecular mechanisms: Recent findings regarding novel
necroptosis regulators. Exp Mol Med. 53:1007–117. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Luedde T, Kaplowitz N and Schwabe RF: Cell
death and cell death responses in liver disease: Mechanisms and
clinical relevance. Gastroenterology. 147:765–83.e4. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Koike A, Hanatani M and Fujimori K:
Pan-caspase inhibitors induce necroptosis via ROS-mediated
activation of mixed lineage kinase domain-like protein and p38 in
classically activated macrophages. Exp Cell Res. 380:171–179. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Horvath C, Young M, Jarabicova I,
Kindernay L, Ferenczyova K, Ravingerova T, Lewis M and Suleiman MS:
Inhibition of cardiac RIP3 mitigates early reperfusion injury and
Calcium-induced mitochondrial swelling without altering necroptotic
signalling. Int J Mol Sci. 22:79832021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Luo Y, Apaijai N, Liao S, Maneechote C,
Chunchai T, Arunsak B, Benjanuwattra J, Yanpiset P, Chattipakorn SC
and Chattipakorn N: Therapeutic potentials of cell death inhibitors
in rats with cardiac ischaemia/reperfusion injury. J Cell Mol Med.
26:2462–2476. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Guo R and Ren J: Alcohol dehydrogenase
accentuates ethanol-induced myocardial dysfunction and
mitochondrial damage in mice: Role of mitochondrial death pathway.
PLoS One. 5:e87572010. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Gao Y, Xu Y, Hua S, Zhou S and Wang K:
ALDH2 attenuates Dox-induced cardiotoxicity by inhibiting cardiac
apoptosis and oxidative stress. Int J Clin Exp Med. 8:6794–6803.
2015.PubMed/NCBI
|
|
121
|
Liu M, Li H, Yang R, Ji D and Xia X:
GSK872 and necrostatin-1 protect retinal ganglion cells against
necroptosis through inhibition of RIP1/RIP3/MLKL pathway in
glutamate-induced retinal excitotoxic model of glaucoma. J
Neuroinflammation. 19:2622022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Shen C, Wang C, Han S, Wang Z, Dong Z,
Zhao X, Wang P, Zhu H, Sun X, Ma X, et al: Aldehyde dehydrogenase 2
deficiency negates chronic low-to-moderate alcohol
consumption-induced cardioprotecion possibly via ROS-dependent
apoptosis and RIP1/RIP3/MLKL-mediated necroptosis. Biochim Biophys
Acta Mol Basis Dis. 1863:1912–1918. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhai X, Wang W, Sun S, Han Y, Li J, Cao S,
Li R, Xu T, Yuan Q, Wang J, et al: 4-Hydroxy-2-nonenal promotes
cardiomyocyte necroptosis via stabilizing receptor-interacting
serine/threonine-protein kinase 1. Front Cell Dev Biol.
9:7217952021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Fang T, Cao R, Wang W, Ye H, Shen L, Li Z,
Hu J and Gao Q: Alterations in necroptosis during ALDH2-mediated
protection against high glucose-induced H9c2 cardiac cell injury.
Mol Med Rep. 18:2807–2815. 2018.PubMed/NCBI
|
|
125
|
Zhang J, Wang R, Xie L, Ren H, Luo D, Yang
Y, Xie H, Shang Z and Liu C: Pharmacological activation of aldehyde
dehydrogenase 2 inhibits ferroptosis via SLC7A11/GPX4 axis to
reduce kidney stone formation. Eur J Pharmacol. 986:1771322025.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Chen J, Hu C, Lu X, Yang X, Zhu M, Ma X
and Yang Y: ALDH2 alleviates inflammation and facilitates
osteogenic differentiation of periodontal ligament stem cells in
periodontitis by blocking ferroptosis via activating Nrf2. Funct
Integr Genomics. 24:1842024. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang Y, Yuan Z, Chai J, Zhu D, Miao X,
Zhou J and Gu X: ALDH2 ameliorates ethanol-induced gastric ulcer
through suppressing NLPR3 inflammasome activation and ferroptosis.
Arch Biochem Biophys. 743:1096212023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Shan G, Bian Y, Yao G, Liang J, Shi H, Hu
Z, Zheng Z, Bi G, Fan H and Zhan C: Targeting ALDH2 to augment
platinum-based chemosensitivity through ferroptosis in lung
adenocarcinoma. Free Radic Biol Med. 224:310–324. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhu ZY, Liu YD, Gong Y, Jin W, Topchiy E,
Turdi S, Gao YF, Culver B, Wang SY, Ge W, et al: Mitochondrial
aldehyde dehydrogenase (ALDH2) rescues cardiac contractile
dysfunction in an APP/PS1 murine model of Alzheimer's disease via
inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol Sin.
43:39–49. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Li W, Yin L, Sun X, Wu J, Dong Z, Hu K,
Sun A and Ge J: Alpha-lipoic acid protects against pressure
overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1
signaling. Cell Death Dis. 11:5992020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ma X, Luo Q, Zhu H, Liu X, Dong Z, Zhang
K, Zou Y, Wu J, Ge J and Sun A: Aldehyde dehydrogenase 2 activation
ameliorates CCl4-induced chronic liver fibrosis in mice by
up-regulating Nrf2/HO-1 antioxidant pathway. J Cell Mol Med.
22:3965–3978. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tao Y, Huang X, Xie Y, Zhou X, He X, Tang
S, Liao M, Chen Y, Tan A, Chen Y, et al: Genome-wide association
and gene-environment interaction study identifies variants in ALDH2
associated with serum ferritin in a Chinese population. Gene.
685:196–201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Chen CH, Budas GR, Churchill EN, Disatnik
MH, Hurley TD and Mochly-Rosen D: Activation of aldehyde
dehydrogenase-2 reduces ischemic damage to the heart. Science.
321:1493–1495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Beretta M, Gorren AC, Wenzl MV, Weis R,
Russwurm M, Koesling D, Schmidt K and Mayer B: Characterization of
the East Asian variant of aldehyde dehydrogenase-2: Bioactivation
of nitroglycerin and effects of Alda-1. J Biol Chem. 285:943–952.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Perez-Miller S, Younus H, Vanam R, Chen
CH, Mochly-Rosen D and Hurley TD: Alda-1 is an agonist and chemical
chaperone for the common human aldehyde dehydrogenase 2 variant.
Nat Struct Mol Biol. 17:159–1564. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Doorn JA, Hurley TD and Petersen DR:
Inhibition of human mitochondrial aldehyde dehydrogenase by
4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol.
19:102–110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Budas GR, Disatnik MH and Mochly-Rosen D:
Aldehyde dehydrogenase 2 in cardiac protection: A new therapeutic
target? Trends Cardiovasc Med. 19:158–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Malyshev Y, Neuzil P, Petru J, Funasako M,
Hala P, Kopriva K, Schneider C, Achyutha A, Vanderper A, Musikantow
D, et al: Nitroglycerin to ameliorate coronary artery spasm during
focal Pulsed-Field ablation for atrial fibrillation. JACC Clin
Electrophysiol. 10:885–896. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Mollace V, Muscoli C, Dagostino C,
Giancotti LA, Gliozzi M, Sacco I, Visalli V, Gratteri S, Palma E,
Malara N, et al: The effect of peroxynitrite decomposition catalyst
MnTBAP on aldehyde dehydrogenase-2 nitration by organic nitrates:
Role in nitrate tolerance. Pharmacol Res. 89:29–35. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Salvemini D, Pistelli A and Mollace V:
Release of nitric oxide from glyceryl trinitrate by captopril but
not enalaprilat: In vitro and in vivo studies. Br J Pharmacol.
109:430–436. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Munzel T and Daiber A: The potential of
aldehyde dehydrogenase 2 as a therapeutic target in cardiovascular
disease. Expert Opin Ther Targets. 22:217–231. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C,
Sheng H, Liu Y, Yu J, Xie Y, et al: Mitochondrial aldehyde
dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the
variation in efficacy of sublingual nitroglycerin. J Clin Invest.
116:506–511. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Nagano T, Ushijima K, Taga N, Takeuchi M,
Kawada MA, Aizawa K, Imai Y and Fujimura A: Influence of the
aldehyde dehydrogenase 2 polymorphism on the vasodilatory effect of
nitroglycerin in infants with congenital heart disease and
pulmonary arterial hypertension. Eur J Clin Pharmacol.
75:1361–1367. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Maiuolo J, Oppedisano F, Carresi C,
Gliozzi M, Musolino V, Macri R, Scarano F, Coppoletta A, Cardamone
A, Bosco F, et al: The generation of nitric oxide from aldehyde
Dehydrogenase-2: The role of dietary nitrates and their implication
in cardiovascular disease management. Int J Mol Sci. 23:154542022.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Marini E, Giorgis M, Leporati M, Rolando
B, Chegaev K, Lazzarato L, Bertinaria M, Vincenti M and Di Stilo A:
Multitarget antioxidant NO-donor organic nitrates: A novel approach
to overcome nitrates tolerance, an ex vivo study. Antioxidants
(Basel). 11:1662022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Chen YR, Nie SD, Shan W, Jiang DJ, Shi RZ,
Zhou Z, Guo R, Zhang Z and Li YJ: Decrease in endogenous CGRP
release in nitroglycerin tolerance: Role of ALDH-2. Eur J
Pharmacol. 571:44–50. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kim EJ, Kim SY, Lee JH, Kim JM, Kim JS,
Byun JI and Koo BN: Effect of isoflurane post-treatment on
tPA-exaggerated brain injury in a rat ischemic stroke model. Korean
J Anesthesiol. 68:281–286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zangrillo A, Lomivorotov VV, Pasyuga VV,
Belletti A, Gazivoda G, Monaco F, Nigro Neto C, Likhvantsev VV,
Bradic N, Lozovskiy A, et al: Effect of volatile anesthetics on
myocardial infarction after coronary artery surgery: A post hoc
analysis of a randomized trial. J Cardiothorac Vasc Anesth.
36:2454–2462. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Yuan J and Fu X: MicroRNA-21 mediates the
protective role of emulsified isoflurane against myocardial
ischemia/reperfusion injury in mice by targeting SPP1. Cell Signal.
86:1100862021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ku HC, Huang CW and Lee SY: Technical
refinement of a bilateral renal Ischemia-reperfusion mouse model
for acute kidney injury research. J Vis Exp. Nov 3–2023.doi:
10.3791/63957. View
Article : Google Scholar : PubMed/NCBI
|
|
151
|
Obeid PCI, Natalini CC and Howell GE:
Exposure to emulsified isoflurane and sevoflurane protects canine
primary hepatocytes against hypoxia-induced apoptosis. Am J Vet
Res. 1–8. 2023.doi: 10.2460/ajvr.23.08.0192 (Epub ahead of print).
PubMed/NCBI
|
|
152
|
Li H and Lang XE: Protein kinase C
signaling pathway involvement in cardioprotection during isoflurane
pretreatment. Mol Med Rep. 11:2683–2688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Cleveland JC Jr, Meldrum DR, Rowland RT,
Banerjee A and Harken AH: Adenosine preconditioning of human
myocardium is dependent upon the ATP-sensitive K+ channel. J Mol
Cell Cardiol. 29:175–182. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhao Z, Shi Q, Guo Q, Peng L, Li X, Rao L
and Li M: Remote ischemic preconditioning can extend the tolerance
to extended drug-coated balloon inflation time by reducing
myocardial damage during percutaneous coronary intervention. Int J
Cardiol. 353:3–8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Amador A, Grande L, Marti J, Deulofeu R,
Miquel R, Sola A, Rodriguez-Laiz G, Ferrer J, Fondevila C, Charco
R, et al: Ischemic pre-conditioning in deceased donor liver
transplantation: A prospective randomized clinical trial. Am J
Transplant. 7:2180–2189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Hardt J, Seyfried S, Brodrecht H, Khalil
L, Buttner S, Herrle F, Reissfelder C and Rahbari NN: Remote
ischemic preconditioning versus sham-control for prevention of
anastomotic leakage after resection for rectal cancer (RIPAL
trial): A pilot randomized controlled, triple-blinded monocenter
trial. Int J Colorectal Dis. 39:652024. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Ueta CB, Campos JC, Albuquerque RPE, Lima
VM, Disatnik MH, Sanchez AB, Chen CH, de Medeiros MHG, Yang W,
Mochly-Rosen D and Ferreira JCB: Cardioprotection induced by a
brief exposure to acetaldehyde: Role of aldehyde dehydrogenase 2.
Cardiovasc Res. 114:1006–1015. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Marino A and Levi R: Salvaging the
ischemic heart: Gi-coupled receptors in mast cells activate a
PKCepsilon/ALDH2 pathway providing Anti-RAS cardioprotection. Curr
Med Chem. 25:4416–4431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Churchill EN, Disatnik MH and Mochly-Rosen
D: Time-dependent and ethanol-induced cardiac protection from
ischemia mediated by mitochondrial translocation of varepsilonPKC
and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol.
46:278–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen CH, Gray MO and Mochly-Rosen D:
Cardioprotection from ischemia by a brief exposure to physiological
levels of ethanol: Role of epsilon protein kinase C. Proc Natl Acad
Sci USA. 96:12784–12789. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Kang PF, Wu WJ, Tang Y, Xuan L, Guan SD,
Tang B, Zhang H, Gao Q and Wang HJ: Activation of ALDH2 with low
concentration of ethanol attenuates myocardial Ischemia/Reperfusion
injury in diabetes rat model. Oxid Med Cell Longev.
2016:61905042016. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Li D, Chen J, Ai Y, Gu X, Li L, Che D,
Jiang Z, Li L, Chen S, Huang H, et al: Estrogen-related hormones
induce apoptosis by stabilizing Schlafen-12 protein turnover. Mol
Cell. 75:1103–16.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Ji E, Jiao T, Shen Y, Xu Y, Sun Y, Cai Z,
Zhang Q and Li J: Molecular mechanism of HSF1-Upregulated ALDH2 by
PKC in ameliorating pressure overload-induced heart failure in
mice. Biomed Res Int. 2020:34816232020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Wang S, Wang L, Qin X, Turdi S, Sun D,
Culver B, Reiter RJ, Wang X, Zhou H and Ren J: ALDH2 contributes to
melatonin-induced protection against APP/PS1 mutation-prompted
cardiac anomalies through cGAS-STING-TBK1-mediated regulation of
mitophagy. Signal Transduct Target Ther. 5:1192020. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Luo G, Huang B, Qiu X, Xiao L, Wang N, Gao
Q, Yang W and Hao L: Resveratrol attenuates excessive ethanol
exposure induced insulin resistance in rats via improving NAD+/NADH
ratio. Mol Nutr Food Res. 612017.doi: 10.1002/mnfr.201700087.
|
|
166
|
Zhang H, Xue L, Li B, Zhang Z and Tao S:
Vitamin D protects against alcohol-induced liver cell injury within
an NRF2-ALDH2 feedback loop. Mol Nutr Food Res. 63:e18010142019.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
He JD and Parker JD: The effect of vitamin
C on nitroglycerin-mediated vasodilation in individuals with and
without the aldehyde dehydrogenase 2 polymorphism. Br J Clin
Pharmacol. 89:2767–2774. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Rodriguez-Gutierrez G, Duthie GG, Wood S,
Morrice P, Nicol F, Reid M, Cantlay LL, Kelder T, Horgan GW,
Fernández-Bolaños Guzmán J and de Roos B: Alperujo extract,
hydroxytyrosol, and 3,4-dihydroxyphenylglycol are bioavailable and
have antioxidant properties in vitamin E-deficient rats-a
proteomics and network analysis approach. Mol Nutr Food Res.
56:1137–1147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Hosoi T, Yamaguchi R, Noji K, Matsuo S,
Baba S, Toyoda K, Suezawa T, Kayano T, Tanaka S and Ozawa K:
Flurbiprofen ameliorated obesity by attenuating leptin resistance
induced by endoplasmic reticulum stress. EMBO Mol Med. 6:335–346.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Balber AE: Concise review: Aldehyde
dehydrogenase bright stem and progenitor cell populations from
normal tissues: Characteristics, activities, and emerging uses in
regenerative medicine. Stem Cells. 29:570–575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Perin EC, Silva GV, Zheng Y, Gahremanpour
A, Canales J, Patel D, Fernandes MR, Keller LH, Quan X, Coulter SA,
et al: Randomized, double-blind pilot study of transendocardial
injection of autologous aldehyde dehydrogenase-bright stem cells in
patients with ischemic heart failure. Am Heart J. 163:415–421.e1.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Perin EC, Murphy M, Cooke JP, Moye L,
Henry TD, Bettencourt J, Gahremanpour A, Leeper N, Anderson RD,
Hiatt WR, et al: Rationale and design for PACE: Patients with
intermittent claudication injected with ALDH bright cells. Am Heart
J. 168:667–673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Perin EC, Murphy MP, March KL, Bolli R,
Loughran J, Yang PC, Leeper NJ, Dalman RL, Alexander J, Henry TD,
et al: Evaluation of cell therapy on exercise performance and limb
perfusion in peripheral artery disease: The CCTRN PACE trial
(Patients with intermittent claudication injected with ALDH bright
cells). Circulation. 135:1417–1428. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sun X, Zhu H, Dong Z, Liu X, Ma X, Han S,
Lu F, Wang P, Qian S, Wang C, et al: Mitochondrial aldehyde
dehydrogenase-2 deficiency compromises therapeutic effect of ALDH
bright cell on peripheral ischemia. Redox Biol. 13:196–206. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Chan W, Taylor AJ, Ellims AH, Lefkovits L,
Wong C, Kingwell BA, Natoli A, Croft KD, Mori T, Kaye DM, et al:
Effect of iron chelation on myocardial infarct size and oxidative
stress in ST-elevation-myocardial infarction. Circ Cardiovasc
Interv. 5:270–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Li W, Feng G, Gauthier JM, Lokshina I,
Higashikubo R, Evans S, Kiertiburanakul S, Lee MP, Supparatpinyo K,
Zhang F, et al: Ferroptotic cell death and TLR4/Trif signaling
initiate neutrophil recruitment after heart transplantation. J Clin
Invest. 129:2293–2304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Liu B, Zhao C, Li H, Chen X, Ding Y and Xu
S: Puerarin protects against heart failure induced by pressure
overload through mitigation of ferroptosis. Biochem Biophys Res
Commun. 497:233–240. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Yu W, An S, Shao T, Xu H, Chen H, Ning J,
Zhou Y and Chai X: Active compounds of herbs ameliorate impaired
cognition in APP/PS1 mouse model of Alzheimer's disease. Aging
(Albany NY). 11:11186–11201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Eleftheriadis T, Pissas G, Filippidis G,
Liakopoulos V and Stefanidis I: Reoxygenation induces reactive
oxygen species production and ferroptosis in renal tubular
epithelial cells by activating aryl hydrocarbon receptor.
Mol Med Rep. 23:412021.PubMed/NCBI
|
|
183
|
Chen K, Xu Z, Liu Y, Wang Z, Li Y, Xu X,
Chen C, Xia T, Liao Q, Yao Y, et al: Irisin protects mitochondria
function during pulmonary ischemia/reperfusion injury. Sci Transl
Med. 9:eaao62982017. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Lu J, Xu F and Lu H: LncRNA PVT1 regulates
ferroptosis through miR-214-mediated TFR1 and p53. Life Sci.
260:1183052020. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Stamenkovic A, O'Hara KA, Nelson DC,
Maddaford TG, Edel AL, Maddaford G, Dibrov E, Aghanoori M,
Kirshenbaum LA, Fernyhough P, et al: Oxidized phosphatidylcholines
trigger ferroptosis in cardiomyocytes during ischemia-reperfusion
injury. Am J Physiol Heart Circ Physiol. 320:H1170–H1184. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H,
Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, et al:
Tau-mediated iron export prevents ferroptotic damage after ischemic
stroke. Mol Psychiatry. 22:1520–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Wu S, Yang J, Sun G, Hu J, Zhang Q, Cai J,
Yuan D, Li H, Hei Z and Yao W: Macrophage extracellular traps
aggravate iron overload-related liver ischaemia/reperfusion injury.
Br J Pharmacol. 178:3783–3796. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Xu Y, Li X, Cheng Y, Yang M and Wang R:
Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary
ischemia-reperfusion. FASEB J. 34:16262–16275. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Yamada N, Karasawa T, Wakiya T, Sadatomo
A, Ito H, Kamata R, Watanabe S, Komada T, Kimura H, Sanada Y, et
al: Iron overload as a risk factor for hepatic Ischemia-Reperfusion
injury in liver transplantation: Potential role of ferroptosis. Am
J Transplant. 20:1606–1618. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Zhou L, Xue X, Hou Q and Dai C: Targeting
ferroptosis attenuates interstitial inflammation and kidney
fibrosis. Kidney Dis (Basel). 8:57–71. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Tu H, Zhou YJ, Tang LJ, Xiong XM, Zhang
XJ, Ali Sheikh MS, Zhang JJ, Luo XJ, Yuan C and Peng J: Combination
of ponatinib with deferoxamine synergistically mitigates ischemic
heart injury via simultaneous prevention of necroptosis and
ferroptosis. Eur J Pharmacol. 898:1739992021. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Shi Y, Han L, Zhang X, Xie L, Pan P and
Chen F: Selenium alleviates cerebral Ischemia/reperfusion injury by
regulating oxidative stress, mitochondrial fusion and ferroptosis.
Neurochem Res. 47:2992–3002. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Lv Z, Wang F, Zhang X, Zhang X, Zhang J
and Liu R: Etomidate attenuates the ferroptosis in myocardial
Ischemia/Reperfusion rat model via Nrf2/HO-1 pathway. Shock.
56:440–449. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Yang J, Sun X, Huang N, Li P, He J, Jiang
L, Zhang X, Han S and Xin H: Entacapone alleviates acute kidney
injury by inhibiting ferroptosis. FASEB J. 36:e223992022.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Chen Y, He W, Wei H, Chang C, Yang L, Meng
J, Long T, Xu Q and Zhang C: Srs11-92, a ferrostatin-1 analog,
improves oxidative stress and neuroinflammation via Nrf2 signal
following cerebral ischemia/reperfusion injury. CNS Neurosci Ther.
29:1667–1677. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Li C and Liu Y: Puerarin reduces cell
damage from cerebral ischemia-reperfusion by inhibiting
ferroptosis. Biochem Biophys Res Commun. 693:1493242024. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Ding Y, Li W, Peng S, Zhou G, Chen S, Wei
Y, Xu J, Gu H, Li J, Liu S and Liu B: Puerarin protects against
myocardial Ischemia/Reperfusion injury by inhibiting ferroptosis.
Biol Pharm Bull. 46:524–532. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Guan X, Li X, Yang X, Yan J, Shi P, Ba L,
Cao Y and Wang P: The neuroprotective effects of carvacrol on
ischemia/reperfusion-induced hippocampal neuronal impairment by
ferroptosis mitigation. Life Sci. 235:1167952019. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M,
Wang L, Zhang Y and Liu X: Rehmannioside A improves cognitive
impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2
and SLC7A11/GPX4 signaling pathway after ischemia. J
Ethnopharmacol. 289:1150212022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Yang T, Liu H, Yang C, Mo H, Wang X, Song
X, Jiang L, Deng P, Chen R, Wu P, et al: Galangin attenuates
myocardial ischemic Reperfusion-Induced ferroptosis by targeting
Nrf2/Gpx4 signaling pathway. Drug Des Devel Ther. 17:2495–2511.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Guo H, Zhu L, Tang P, Chen D, Li Y, Li J
and Bao C: Carthamin yellow improves cerebral ischemia-reperfusion
injury by attenuating inflammation and ferroptosis in rats. Int J
Mol Med. 47:522021. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Yuan Y, Zhai Y, Chen J, Xu X and Wang H:
Kaempferol ameliorates Oxygen-glucose
deprivation/reoxygenation-induced neuronal ferroptosis by
activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules. 11:9232021.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Lin JH, Yang KT, Ting PC, Luo YP, Lin DJ,
Wang YS and Chang JC: Gossypol acetic acid attenuates cardiac
ischemia/reperfusion injury in rats via an antiferroptotic
mechanism. Biomolecules. 11:16672021. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Du YW, Li XK, Wang TT, Zhou L, Li HR, Feng
L, Ma H and Liu HB: Cyanidin-3-glucoside inhibits ferroptosis in
renal tubular cells after ischemia/reperfusion injury via the AMPK
pathway. Mol Med. 29:422023. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X,
Huang Z, Lin M, Wu H and Xu D: Naringenin alleviates myocardial
ischemia/reperfusion injury by regulating the nuclear
factor-erythroid factor 2-related factor 2 (Nrf2)/System
xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis.
Bioengineered. 12:10924–10934. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Li T, Tan Y, Ouyang S, He J and Liu L:
Resveratrol protects against myocardial ischemia-reperfusion injury
via attenuating ferroptosis. Gene. 808:1459682022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Zhu H, Huang J, Chen Y, Li X, Wen J, Tian
M, Ren J, Zhou L and Yang Q: Resveratrol pretreatment protects
neurons from oxygen-glucose deprivation/reoxygenation and ischemic
injury through inhibiting ferroptosis. Biosci Biotechnol Biochem.
86:704–716. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Wang X, Shen T, Lian J, Deng K, Qu C, Li
E, Li G, Ren Y, Wang Z, Jiang Z, et al: Resveratrol reduces
ROS-induced ferroptosis by activating SIRT3 and compensating the
GSH/GPX4 pathway. Mol Med. 29:1372023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Zhang J, Bi J, Ren Y, Du Z, Li T, Wang T,
Zhang L, Wang M, Wei S, Lv Y and Wu R: Involvement of GPX4 in
irisin's protection against ischemia reperfusion-induced acute
kidney injury. J Cell Physiol. 236:931–945. 2021. View Article : Google Scholar : PubMed/NCBI
|