Sepsis is a serious infectious disease caused by the
invasion of bacteria, viruses, fungi and other pathogens into human
tissues (1). In severe cases,
sepsis can lead to systemic inflammatory response syndrome,
multiple organ failure and mortality (2–4).
Sepsis is a global health problem and, according to a previous
study, in 2017, there were an estimated 48.9 million cases of
sepsis worldwide, and nearly 11 million sepsis-related mortalities
were reported, which accounted for 19.7% of the total global
mortality (5). In the United
States, >1.7 million adults develop sepsis each year, and sepsis
results in mortality of nearly >350,000 adults annually
(6). In China, from 2017 to 2019,
4.8 to 6.1 million patients were hospitalized with sepsis each
year, and there were ~800,000 sepsis-related mortalities, with
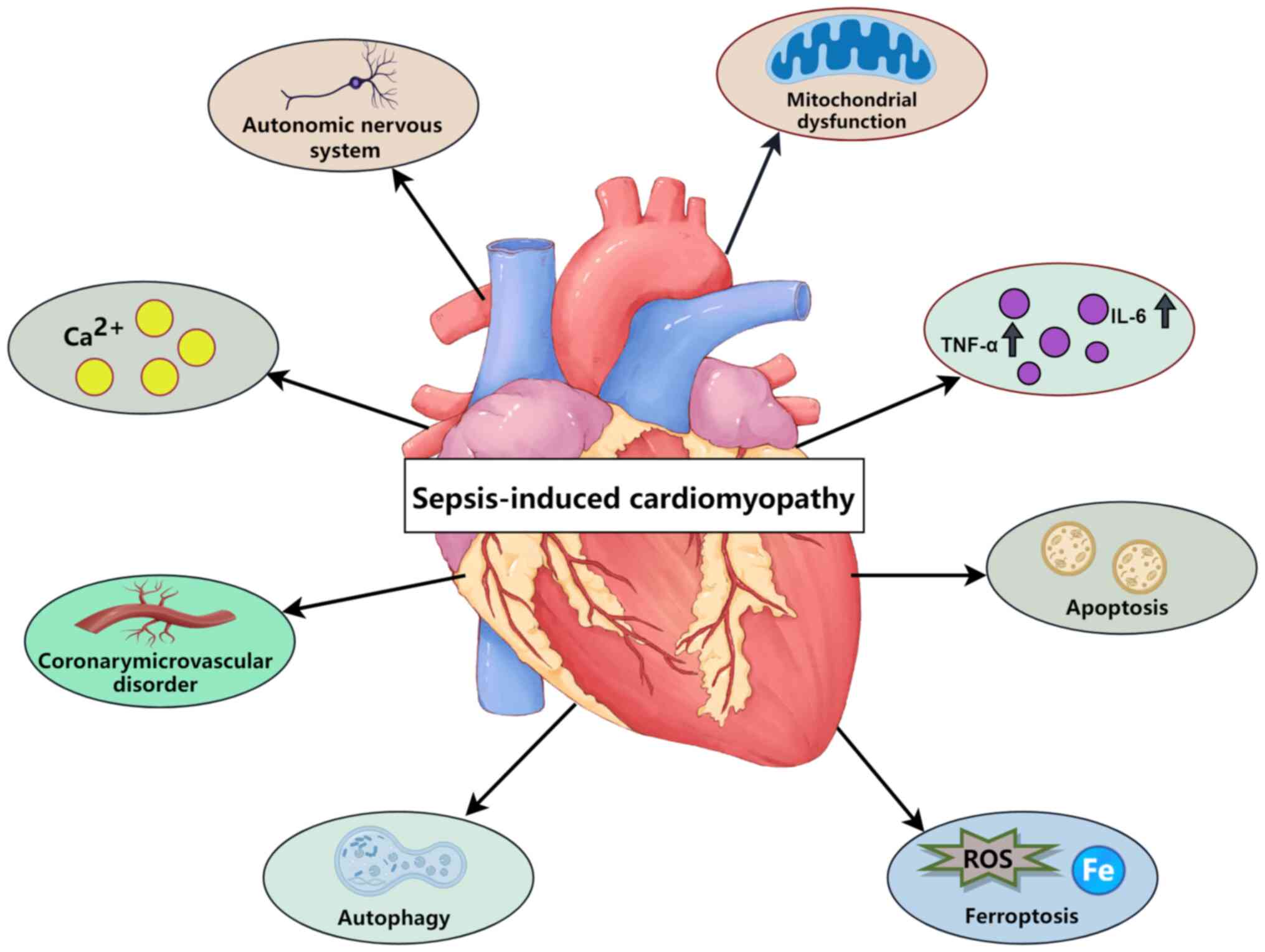
complications occurring in >70% of cases (7). Sepsis-induced cardiomyopathy, a
common and serious complication of sepsis, is a key cause of
mortality in critically serious patients in the intensive care
unit, and can increase the mortality rate of sepsis in patients by
two to three fold (8). The
pathogenesis of SIC is complex and may be associated with various
factors, such as inflammation, apoptosis and energy metabolism
disorders (9). However, the
specific molecular mechanisms are still unclear, and further
research is required.
At the organelle level, mitochondria are closely
associated with the occurrence of ferroptosis (12). Excess iron in cells can be absorbed
by mitochondria, promoting the Fenton reaction to produce reactive
oxygen species (ROS) that cause mitochondrial lipid peroxidation
and damage, thus affecting cellular energy production (13,14).
Mitochondria are abundant in cardiomyocytes and damage to
mitochondria seriously effects their normal function (15).
In previous years, numerous studies have revealed
that ferroptosis has a key role in the occurrence of SIC (16,17).
Therefore, further research on ferroptosis may be beneficial for
the diagnosis, treatment and prognosis of SIC. The present review
briefly describes the molecular mechanisms of ferroptosis.
Additionally, the present review focuses on the research progress
of the possible mechanisms of action in SIC to provide new ideas
for the treatment of SIC in the future.
SIC has not been clearly defined since it was first
proposed and its complex pathophysiological processes hinder
diagnosis. New advances in cardiac imaging have increased the
uncertainty in defining SIC; moreover, the potential for myocardial
dysfunction and the need for acute intervention adds further
complexity (18). A 2019 review
proposed that SIC is an acute cardiac dysfunction caused by sepsis,
which is a reversible complication. The clinical symptoms of SIC
mainly manifest as ventricular dilatation, reduced ventricular
contractility and/or right and left ventricular dysfunction, and a
reduced response to volume infusion (19). These symptoms are largely
associated with cardiac function and hemodynamic changes (20). Wang et al (21) proposed using echocardiographic
changes to explain the possible pathophysiological mechanism of
cardiac function changes in SIC. To date, although there have been
numerous studies and reviews of SIC (16,22–24),
the understanding of SIC is still insufficient, and thus there is
an unmet clinical need in the diagnosis and treatment of SIC. The
present review provides a brief overview of current research on the
mechanisms of SIC (Fig. 1).
SIC is associated with an excessive inflammatory
response, through which the release of inflammatory mediators is a
key factor. Studies have revealed that in patients with sepsis, the
expression of pro-inflammatory factors, such as tumor necrosis
factor-α, IL-1β and IL-6 are increased (8,25,26).
These inflammatory factors are mainly mediated by Toll-like
receptor-4 to recognize different pathogen-associated molecular
patterns and damage-associated molecular patterns (27). Toll-like receptor-4 activates
intracellular signaling pathways such as the MyD88-dependent
pathway (28). This activation
leads to the translocation of NF-κB into the nucleus, where it
promotes the transcription of target genes, including
pro-inflammatory cytokines and chemokines which attract and
activate inflammatory cells such as macrophages and neutrophils
(29). Macrophages, upon
activation, secrete more pro-inflammatory cytokines, creating a
positive feedback loop that exacerbates the inflammatory response
(30). Neutrophils, attracted to
the site of injury, release ROS and proteases, which directly
damage cardiomyocytes, compromising the integrity of the cell
membrane and leading to cell content leakage (31). This direct damage, along with
metabolic disturbances and altered gene expression in
cardiomyocytes induced by the inflammatory response, results in
necrosis and apoptosis of cardiomyocytes (32). Additionally, the inflammatory
response stimulates the activation and proliferation of cardiac
fibroblasts, leading to myocardial fibrosis, which replaces normal
myocardial tissue and impairs cardiac function (33). These above processes eventually
lead to myocardial damage and dysfunction.
Mitochondrial dysfunction is a key characteristic of
SIC. The main role of mitochondria is to produce adenosine
triphosphate (ATP) but they also have roles in other cellular
functions, such as calcium homeostasis, body temperature
regulation, cell signal transduction and apoptosis (34). The manifestations of mitochondrial
dysfunction are complex and diverse in SIC, mainly in the electron
transport chain and oxidative phosphorylation abnormalities,
mitochondrial structure abnormalities, oxidative and nitrite
stress, mitochondrial calcium overload, mitochondrial autophagy
dysregulation, and mitochondrial dynamics imbalance (35–37).
The electron transport chain within mitochondria is
a pivotal component of cellular respiration and energy generation.
During SIC, disruptions of this chain lead to reduced ATP
synthesis, thereby impairing the regular operation of
cardiomyocytes (38). Deviations
in mitochondrial morphology and structure, such as swelling,
shortening and the loss of cristae, can also compromise
mitochondrial function. Furthermore, heightened levels of ROS and
reactive nitrogen species generated by mitochondria can initiate
oxidative and nitrosative stress, exacerbating damage to both
mitochondria and cardiomyocytes (39). Calcium has a key role in
mitochondrial function and is a primary regulator of oxidative
phosphorylation (40). In SIC,
elevated levels of calcium ions within the mitochondria contribute
to reduced mitochondrial antioxidant capacity and heightened
production of ROS (41).
Additionally, autophagy, which is responsible for the elimination
of damaged mitochondria, may become dysregulated, resulting in the
accumulation of dysfunctional mitochondria and thus cellular
malfunction (42).
Mitochondrial division and fusion are key processes
that preserve the dynamic equilibrium of the mitochondrial network.
When this equilibrium is disrupted, it can adversely affect
mitochondrial function and distribution. Failure of the
mitochondrial antioxidant system increases susceptibility of the
body to inflammation, immune response, hormone metabolism and
bioenergetic response activation (43). This failure mainly leads to
increased production of ROS, which can damage mitochondrial
components (44). The accumulation
of ROS triggers a series of detrimental processes: It activates
inflammatory pathways, causing the release of pro-inflammatory
cytokines and initiating an inflammatory response; stimulates the
immune system, potentially leading to an overactive immune
response; disrupts hormone synthesis and metabolism, causing
hormonal imbalances; and reduces ATP production efficiency,
impairing the cell's ability to meet its energy demands and
affecting various bioenergetic processes (45,46).
These processes create a vicious cycle, where initial mitochondrial
dysfunction leads to oxidative stress, which further exacerbates
mitochondrial issues and increases ROS production, ultimately
resulting in an irreversible condition of oxidative stress and
mitochondrial dysfunction that impacts the cell's overall health
and function (47).
Cell death is another key feature of SIC. All types
of damage can result in irreversible damage of myocardial cells and
promote the progression of cardiac dysfunction (48). Most cardiomyocyte death is
accomplished through regulatory molecular pathways, including
autophagy and apoptosis, pyroptosis, ferroptosis and necrosis.
These forms of regulatory cell death are the main characteristics
of the pathogenesis of SIC (49).
Autophagy is a cellular process that involves the
recycling of materials, and is typically initiated during nutrient
deprivation or other stressful conditions to aid in the elimination
of damaged proteins and organelles (50). Apoptosis is an orderly process of
cellular demise, often initiated by internal signals. In SIC,
autophagy can be hyperactivated or suppressed and inflammatory
mediators along with oxidative stress can induce apoptosis, both of
which contribute to the death of cardiomyocytes (51). Pyroptosis, another mode of cell
death, is triggered by inflammatory vesicles and is characterized
by the discharge of substantial quantities of proinflammatory
cytokines, such as IL-18 and IL-1β. Pyroptosis is particularly
pronounced in SIC as it can amplify inflammatory responses,
resulting in tissue damage and organ dysfunction (52). Ferroptosis, a more recently
discovered form of cell death, is closely associated with
intracellular iron metabolism and lipid peroxidation. During
sepsis, ferroptosis may be initiated by disruptions in iron
metabolism and heightened oxidative stress, leading to damage and
death of cardiomyocytes (53).
Additionally, necrosis is an uncontrolled form of cell death that
can interact with other forms of cell death, ultimately
contributing to myocardial tissue damage (9). Understanding the regulatory
mechanisms underlying these cell death pathways is key for the
advancement of novel therapeutic strategies for SIC.
In addition to the aforementioned mechanisms,
calcium dysregulation, autonomic nervous system dysfunction and
coronary microvascular disorders may be involved in SIC (54). Disturbances in intracellular
calcium homeostasis have been studied in septic hearts (55). Most models of sepsis show a
decrease in cytosolic calcium transients, the difference between
systolic and diastolic calcium, which may be associated with an
increase in diastolic cytoplasmic calcium and a decrease in
sarcoplasmic reticulum calcium content (8,56).
In sepsis, the hypothalamic-pituitary-adrenal axis and the
sympathetic nervous system are the two main pathways of
neuromodulation (57).
Hyperactivation of the sympathetic nervous system and elevated
levels of circulating endogenous catecholamines ultimately
culminate in the desensitization of adrenergic receptors, which in
turn affects cardiac function (58). Alterations in the coronary
microvasculature may affect myocardial blood flow and oxygen
supply. This occurs mainly due to disruption of the glycocalyx in
the endothelium leading to vascular leakage, coagulation and
inflammation, with the heterogeneous microvascular flow and
myocardial edema (59).
The occurrence and development of numerous diseases,
such as various types of cancer, neurodegenerative diseases,
diseases involving tissue or organ damage and inflammation, and
infectious diseases, are inseparable from ferroptosis (60). The morphological and mechanistic
characteristics of ferroptosis are different from those of
traditional cell death. In terms of morphology, ferroptosis mainly
manifests as a reduction in the levels of mitochondria, an increase
in mitochondrial membrane density, a reduction in the number of
mitochondrial cristae and rupture of the mitochondrial outer
membrane. However, for all cells undergoing ferroptosis, there is
no considerable association between change in nuclear size or
nuclear chromatin with ferroptosis (61). It is necessary for particular
lipids to experience oxidation, and concurrently, the physiological
defenses that inhibit the aggregation of oxidized lipids must be
dysfunctional. The mechanism of ferroptosis is complex and varied
but it is essentially an imbalance between oxidation and
antioxidant systems, resulting in cellular lipid peroxidation
(62). The main pathways resulting
in ferroptosis are discussed in the present review.
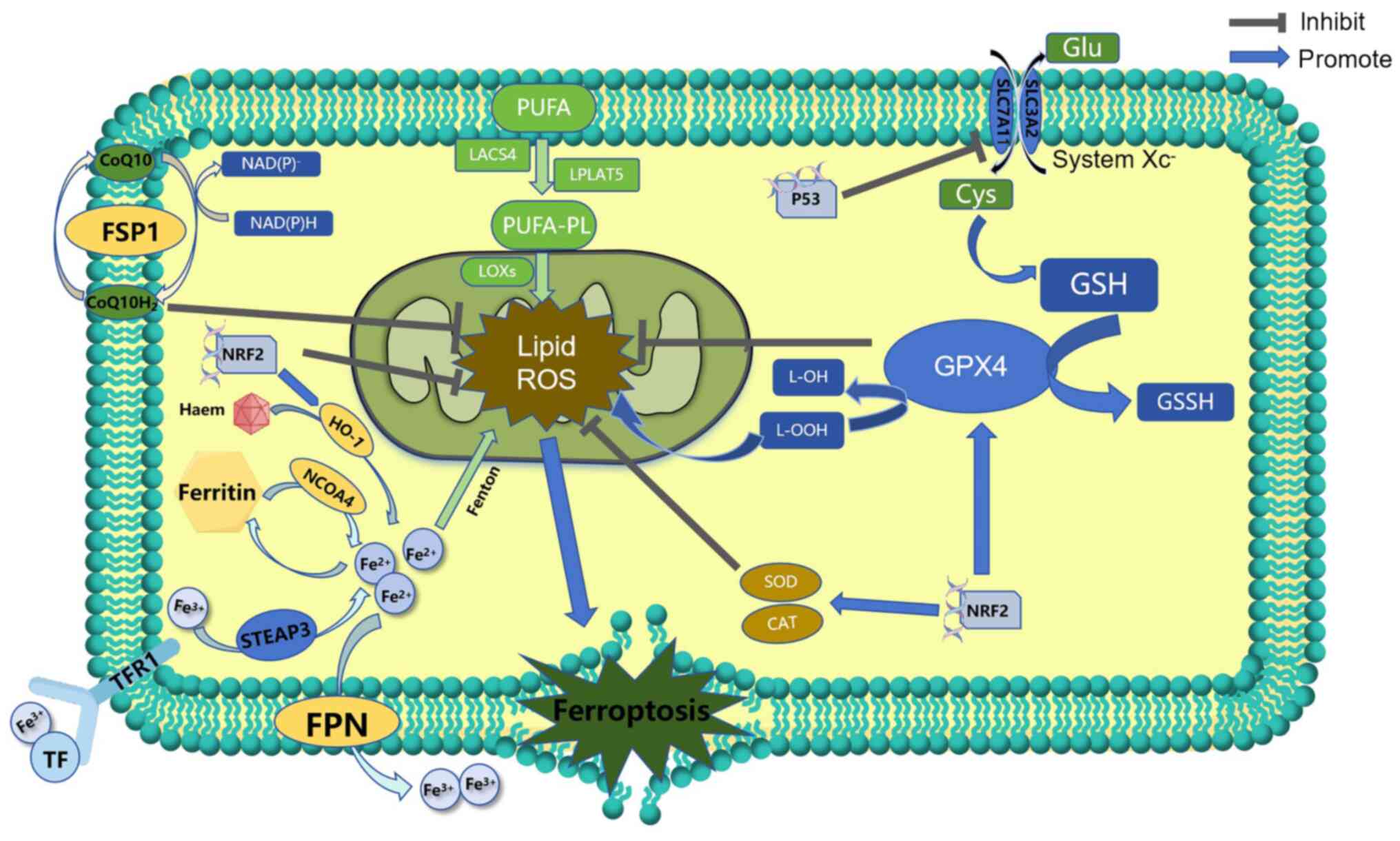
Cystine/glutamate antiporter is a transmembrane
amino acid transporter, also known as system Xc−, which
is distributed in phospholipid bilayers in certain cell types, such
as neurons, immune cells and cancer cells (63). The transporter protein, xCT, in the
transporter is formed of two chains, solute carrier family 7 member
11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2),
connected by disulfide bonds (64). Glutathione (GSH) is a key
antioxidant substance in cells, and cystine is one of the raw
materials needed for its synthesis. By inhibiting activation of
system Xc−, the absorption of cystine is reduced and the
synthesis of GSH is diminished. As a result, the antioxidant
capacity of cells decreases and a large amount of ROS accumulates,
leading to oxidative damage and ferroptosis (65). Thus, ferroptosis can occur by
reducing or depleting GSH: The P53 gene can affect GSH synthesis
and activity by downregulating the expression of SLC7A11 (66). Beclin1 can directly bind to SLC7A11
and contribute to the depletion of GSH and the level of lipid
peroxidation (67). The
ATP-binding cassette family of transporters of the multidrug
resistance protein 1 can enable the release of GSH into the
extracellular space, leading to GSH depletion (68). These result in ROS accumulation
(Fig. 2). The ferroptosis inducer
erastin acts by restraining expression of SLC7A11 (69).
Disturbances in iron metabolism can also trigger
ferroptosis. Iron is a vital trace element in the body (77). Therefore, abnormal iron
distribution and content affect normal physiological processes,
such as oxygen transport, DNA synthesis and ATP production
(78). Excessive absorption or
decreased use of iron in cells leads to an imbalance in iron ion
concentrations for various reasons, and this results in iron
overload. Excess iron then undergoes the Fenton reaction, and
hydrogen peroxide produces hydroxyl free radicals and oxygen free
radicals. These react with lipid components in cells and this
generates a large number of lipid ROS eventually resulting in
ferroptosis (Fig. 2) (60,79).
Cellular iron homeostasis in organisms is tightly
regulated. Extracellular iron ions, usually in the form of
trivalent iron, bind to transferrin and enter the cell via receptor
for transferrin-1 (Fig. 2)
(80). After the reduction of
trivalent iron to divalent iron by the metalloreductase
six-transmembrane epithelial antigen of the prostate 3, various
iron complexes or storage iron are formed. When the complex becomes
saturated, excess iron divalent accumulates in the cell and form an
unstable iron pool, which is known as iron overload (81). Therefore, the regulation of iron
metabolism for ferroptosis is dependent on the regulation of the
capacity of the unstable iron pool. Heat shock protein β-1 can
reduce iron uptake and control capacity of the iron pool by
inhibiting TFR-1 expression levels (82). In addition, ferroportin and
ferritin transfer protein, Prominin 2, can transport iron ions and
ferritin to the extracellular compartment through each group of
pathways, respectively, and their reduced expression levels have
been shown to promote ferroptosis (83,84).
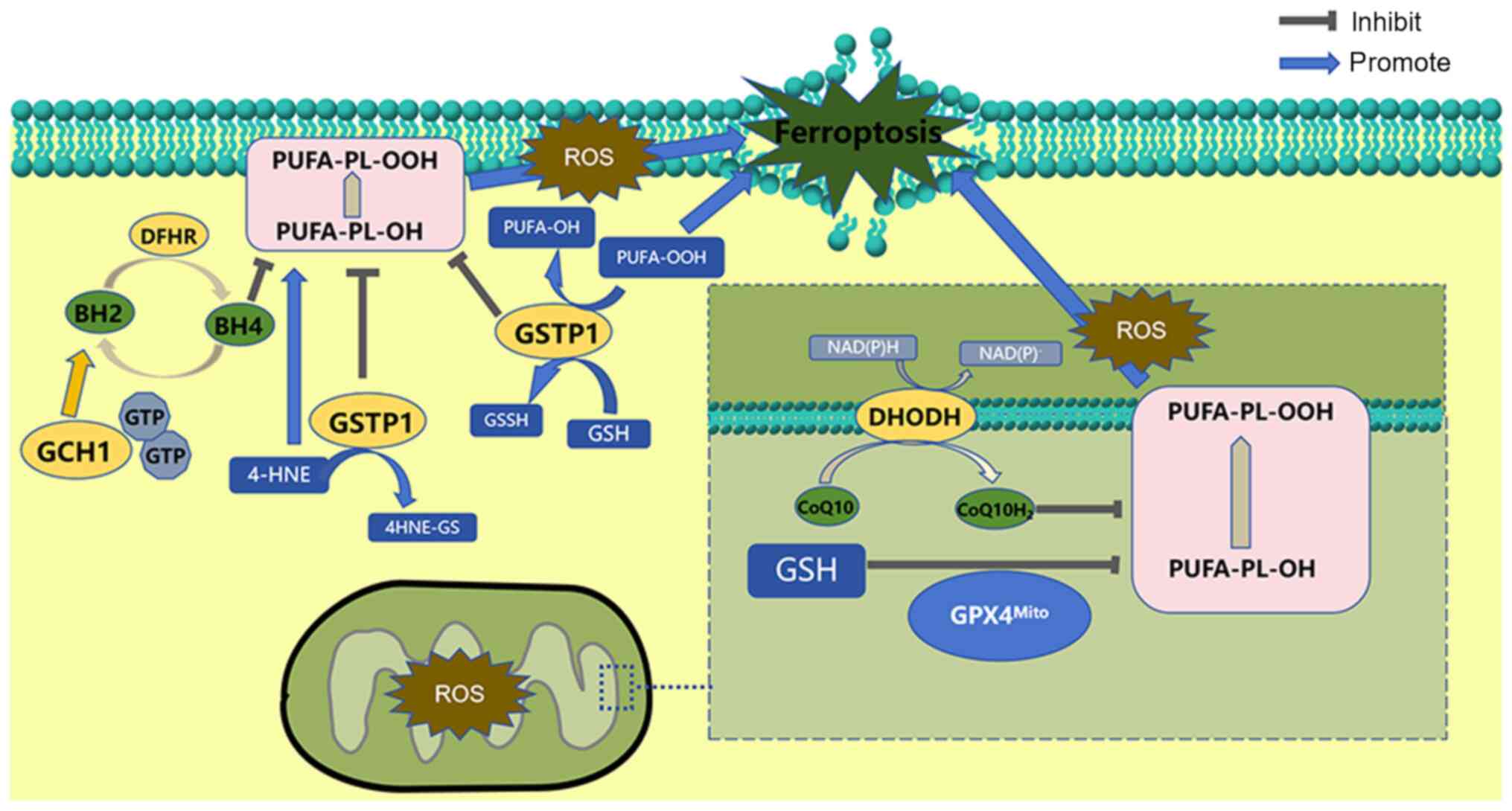
The other is dihydroorotate dehydrogenase (DOHDH).
As a flavin-dependent enzyme located in the inner mitochondrial
membrane, DOHDH reduces ubiquinone CoQ10 to dihydro-ubiquinone,
which traps oxidants in the cell membrane to prevent lipid
peroxidation and thus inhibit ferroptosis (91). DOHDH has a similar role to FSP1 on
the outer mitochondrial membrane (92). On this basis, a subsequent study
has revealed that DOHDH inhibitors make cells more sensitive to
ferroptosis by inhibiting FSP1 but not DHODH (93) (Fig.
3).
A recent study has found that membrane-bound
O-acyltransferase domains 1,2 (MBOAT1 and 2) can reduce
intracellular phosphatidylethanolamine polyunsaturated fatty acids.
And since phosphatidylethanolamine polyunsaturated fatty acid is
the preferred substrate for phospholipid peroxidation and a key
determinant of ferroptosis sensitivity, MBOAT1 and MBOAT2 can
effectively inhibit ferroptosis through a phospholipid remodeling
mechanism (94). MBOAT1 and MBOAT2
are transcriptionally upregulated by the estrogen receptor (ER) and
the androgen receptor (AR), respectively. The combination of ER or
AR antagonists with ferroptosis induction can significantly inhibit
the growth of ER-positive breast cancer or AR-positive prostate
cancer. This finding provides new ideas for the treatment of
cancers with specific genetic backgrounds (94,95).
In addition, GTP cyclohydrolase 1 (GTPCH1) and GSH transferase pi
inhibit ferroptosis independently (Fig. 3) (96,97).
Lipid metabolism is an essential process of
ferroptosis. Polyunsaturated fatty acids in cell membranes or
organelles generate lipid peroxides in response to oxygen free
radicals. When an antioxidant imbalance occurs in the body, the
accumulated lipid peroxides eventually destroy the structure and
function of the membranes, leading to cell damage and death
(98). Furthermore, lipoxygenases
and cyclooxygenases, such as long-chain acyl-CoA synthetase 4,
lysophospholipid acyltransferase 5 and lipoxygenase, facilitate
lipid peroxidation and ferroptosis (99).
Ferroptosis is a common feature of several
cardiovascular diseases and a key process in mediating the
pathogenesis and progression of heart diseases (100,101). These diseases include
atherosclerosis, myocardial ischemia-reperfusion injury, SIC,
drug-induced heart failure, arrhythmia and diabetic cardiomyopathy
(102). SIC is a cardiac disease
caused by sepsis, presenting significant difficulties in both
clinical diagnosis and treatment (9). By summarizing the research on
ferroptosis, progress can be made in the treatment of patients with
SIC.
Mitochondria are characterized as the energy
factories of eukaryotic cells and have a variety of important
functions in different physiological cellular processes (103). Mitochondrial dysfunction is a
catalyst for a variety of diseases with widespread cell death
(104). During SIC, mitochondrial
dysfunction in cardiomyocytes results from heightened oxidative
stress due to the release of inflammatory factors. A previous study
has shown that morphological changes due to mitochondrial damage in
the lipopolysaccharide (LPS)-induced myocardium are consistent with
mitochondrial changes characterized by ferroptosis (105). Therefore, mitochondrial
homeostasis in SIC can be maintained by targeting ferroptosis
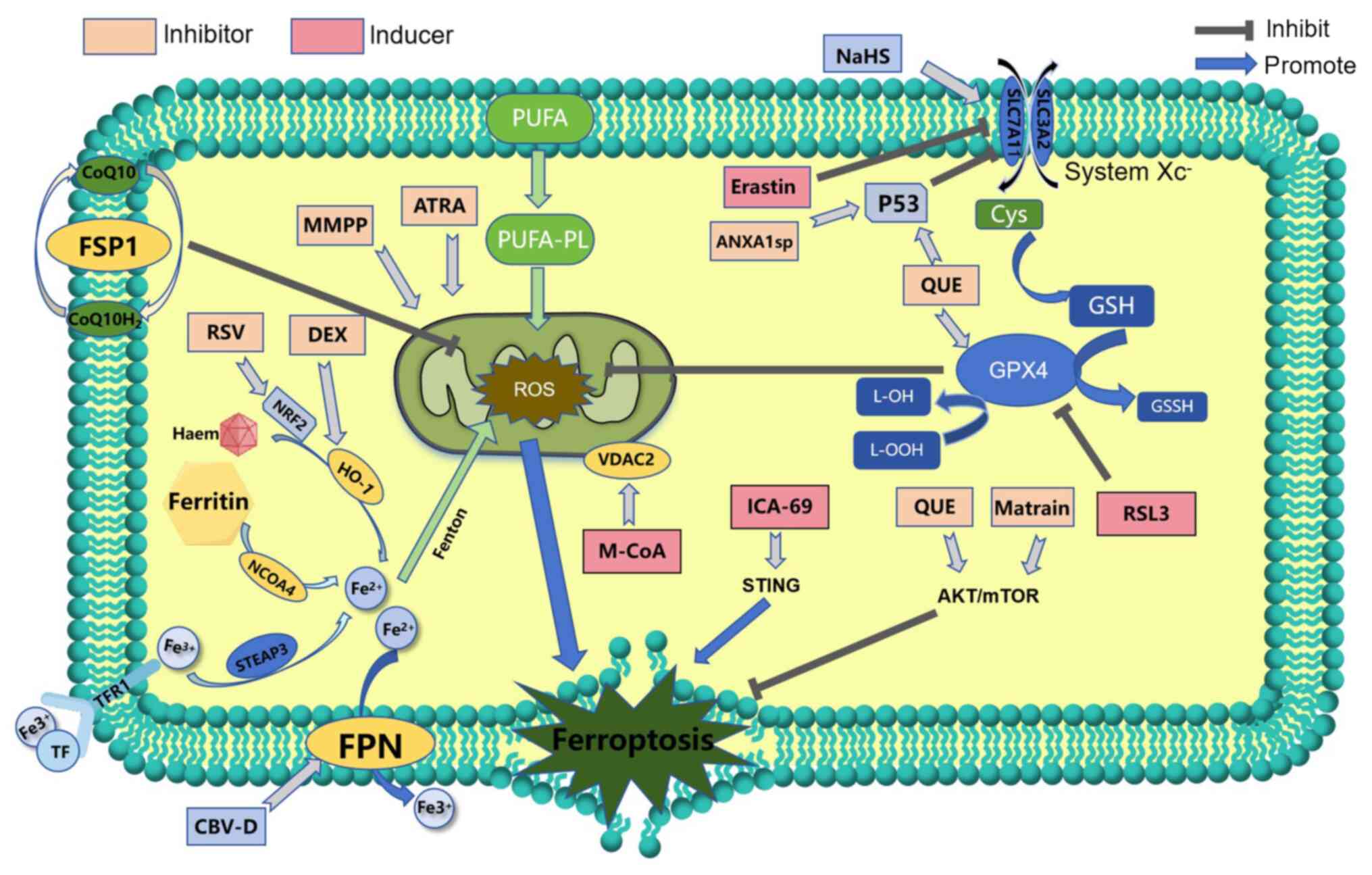
(Fig. 4) (17,35).
Inhibition of xCT protein or GPX4 leads to
ferroptosis, which provides a possible therapeutic target for the
treatment of septic myocardial injury (Fig. 4). Methyltransferase-like 3 (METTL3)
is a core catalytic subunit for RNA N6-methyladenosine (m6A)
modification. METTL3 has a key role in several biological
processes, such as embryonic development, immune responses and the
occurrence and progression of tumors (111). METTL3-mediated m6A methylation
causes the mRNA of SLC7A11 to have increased levels of methylation.
YTH domain family member 2 directly binds to the m6A modification
site to mediate mRNA degradation and promote the decay of SLC7A11
mRNA, thus upregulating ferroptosis in myocardial injuries caused
by sepsis (112). The study by
Cao et al (113) found
that Beclin 1, which is an endogenous SLC7A11 binding protein, can
regulate ferroptosis. Sodium hydrosulfide can inhibit the
phosphorylation of Beclin 1, increase SLC7A11 and GPX4 expression
levels, and reduce the incidence of cardiac ferroptosis, thus
protecting against sepsis-induced cardiomyocyte injury (113). Furthermore, YiQiFuMai injection
has been found to ameliorate SIC by inhibiting ferroptosis via the
xCT/GPX4 axis (Fig. 4) (114).
Previous studies have shown that ferroptosis is
affected by regulation of iron metabolism in SIC. Ferritin
autophagy mediated by nuclear receptor coactivator 4 promotes
ferroptosis by controlling intracellular iron homeostasis (9,120,121). On this basis, expression of
intracellular nuclear receptor coactivator 4 and iron are increased
in an LPS-induced cardiac injury model (122). This is largely due to the fact
that nuclear receptor coactivator 4 can directly interact with
ferritin and degrade it in a ferritin autophagy-dependent manner,
subsequently releasing a large amount of iron, which leads to iron
accumulation (123). Excess iron
in the cytoplasm further stimulates the expression of ferritin on
the mitochondrial membrane, which in turn transports cytoplasmic
iron into the mitochondria, stimulating mitochondrial ROS
production and ferroptosis. These findings suggests that ferritin
autophagy-mediated ferroptosis is involved in SIC (Fig. 4) (124).
Heme oxygenase 1 (HO-1) is an intracellular enzyme
that catalyzes the production of ferrous iron, carbon monoxide and
bilirubin from heme. HO-1 is one of the downstream targets of
nuclear factor erythroid 2-related factor 2 (Nrf2) (Fig. 4). Nrf2 has antioxidant roles, and
increased HO-1 expression reduces oxidative damage and inflammation
(125). HO-1 is also involved in
scavenging free radicals and modulating immune responses. However,
overactivation of HO-1 can contribute to the accumulation of a
large amount of iron in the cytoplasm, promoting ROS production and
ultimately ferroptosis (126,127). Dexmedetomidine, which is an
α2-adrenergic receptor agonist, can diminish ferroptosis
by restricting HO-1 overexpression, decreasing iron concentrations
and increasing levels of antioxidants such as GPX4, which defend
against sepsis-induced cardiomyocyte injury (128). Recombinant ferroportin, which is
located in the cell membrane, has a key role in iron metabolism.
Ferroportin is able to transport iron ions from the intracellular
to the extracellular compartments, thus modulating the distribution
and use of iron in the body (129). Cyclovirobuxine D increases the
upregulation of ferroportin-1, which reduces LPS-induced iron
overload in the cytoplasm, thereby alleviating lipid peroxidation
and ferroptosis in sepsis (130).
Other drugs and mechanisms have been identified that
may be implicated in the development of ferroptosis in SIC
(Fig. 4). Islet cell autoantigen
69, which is a molecule that regulates inflammatory and immune
responses in a variety of diseases, is highly expressed in
cardiomyocytes in wild-type mice with LPS-induced sepsis (131). This molecule triggers the
production of stimulator of interferon genes, further contributing
to lipid peroxidation and ferroptosis in cardiomyocytes.
Transmembrane protein 43 is a quaternary
transmembrane protein that is mainly localized in the endoplasmic
reticulum and inner nuclear membrane and is associated with
cardiovascular diseases (132).
The study by Chen et al (133) revealed that transmembrane protein
43 could be used as a beneficial factor to effectively inhibit
ferroptosis of cardiomyocytes and alleviate myocardial injury.
Transient receptor potential melastatin 7, a
transmembrane protein with the dual functions of cation channel and
kinase activity, can mediate endoplasmic reticulum stress and
ferroptosis. It may be a potential strategy for the treatment of
SIC (134).
Currently, biological agents and their synthetics
are available in a variety of applications and are gradually being
used to examine ferroptosis. The study by Jiang et al
(135) designed cerium dioxide
nanozymes ligated with curcumin by self-assembly of human serum
albumin. The formed cerium dioxide nanozymes ligated with curcumin
acquired superoxide dismutase-like and catalase-like activities of
cerium dioxide nanozymes, which could scavenge ROS (135). This study also reversed
ferroptosis induced by RSL3 and showed an inhibitory effect on
ferroptosis. Additionally, it revealed a reduction in
pro-inflammatory factor release, which substantially relieved
myocardial injury and restored cardiac function. The study by Jiao
et al (136) revealed that
platelet-rich plasma not only reduces inflammation, but also
mitigates oxidative stress and ferroptosis by modulating the
AKT/mTOR signaling pathway.
Numerous studies have investigated
ferroptosis-related genes in search of potential diagnostic and
therapeutic targets (142,143).
The study by Huang et al (144) revealed that the Lcn2 gene
is associated with iron metabolism through bioinformatics analysis.
This study also revealed that knockdown of LncRNA Lcn2-204 has a
cardioprotective effect through the inhibition of iron overload and
ferroptosis in SIC. They study by Song et al (145) showed that the cuproptosis- and
ferroptosis-related genes POR, SLC7A5 and STAT3 are
associated with SIC, and could also be used to predict therapeutic
agents against these targets. The study by Zou et al
(146) identified eight key
ferroptosis-related genes and ferroptosis signatures through the
Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) database. The study
concluded that the aforementioned ferroptosis-related genes exhibit
excellent diagnostic ability for SIC and that some of these genes
are associated with the prognosis of SIC and have potential as
therapeutic drugs. The study by Lin et al (147) screened the expression of six
circadian genes to reveal that the Bmal gene inhibits the
progression of ferroptosis in SIC. Furthermore, Hmox1 and
SLC7A11 are target genes for ferroptosis in sepsis-induced
myocardial injury (148).
In summary, ferroptosis is key in the development of
SIC, but several questions remain to be answered in this field of
research. Despite the well-known regulatory pathways of
ferroptosis, such as the GSH/GPX4 regulatory pathway, the
FSP1/CoQ10 pathway, and iron metabolism and lipid metabolism,
whether there are other regulatory mechanisms is unclear.
Additionally, there is a lack of specific markers for the
occurrence of ferroptosis in SIC.
Currently available established models of SIC are
limited to animal and cellular experiments. For instance, Lu et
al (149) established a mouse
model of SIC by cecum ligation and puncture, including Nrf2
knockout (Nrf2+/−) and wild-type (Nrf2+/+)
mouse, demonstrating that Daohe Chengqi decoction can inhibit
ferroptosis and alleviate SIC. Another Lu et al (150) created a model of SIC by injecting
Sprague-Dawley rats with LPS, showing that nicorandil, a drug used
in the clinical treatment of cardiac diseases such as acute
myocardial infarction and acute heart failure (151), can modulate ferroptosis and
protect the myocardium. These models represent the promotion or
inhibition of ferroptosis by drugs and chemical compounds, and
provide new therapeutic ideas and targets for intervention.
However, they are not supported by clinical data and have not been
truly translated into real-world clinical problems. Most research
has only demonstrated an effect on ferroptosis in models of
LPS-induced cardiomyopathy, and no reliable mechanism or pathway
has been identified to date.
Furthermore, the subcellular organelles that drive
ferroptosis are also essential in SIC. Some studies have
investigated the role of subcellular organelles associated with
ferroptosis in sepsis-related organ injuries, such as the lung
(152,153). However, to the best of our
knowledge, few studies have focused the heart. Therefore, further
investigation is warranted.
As research progresses, the mechanisms and
therapeutic targets associated with ferroptosis in SIC are being
revealed at the genetic level. Therefore, bioinformatics analysis,
technology and genomics should be the priority direction for future
research. Research on this topic can be more convincing and
influential by applying the central law, from transcription and
translation of genes to the establishment of animal and cellular
models for validation. In conclusion, ferroptosis has broad
developmental prospects, and it is expected to be a new approach
for treating SIC by precisely manipulating its molecular
mechanism.
Not applicable.
This work was supported by The Beijing Natural Science
Foundation (grant no. 7232126), The Special Scientific Research
Project of Beijing Critical Care Ultrasound Research Association
(grant no. 2023-CCUSG-A-03) and The Medical Health Research Project
of Yichang (grant. no. A24-2-011).
Not applicable.
DW drafted the manuscript and prepared the figures.
XQ revised the manuscript and participated in the design of the
figures. ZZ designed the present study and revised the manuscript.
GZ designed the present study and revised the manuscript. All
authors have read and approved the final manuscript. All authors
are responsible for all aspects of the work and approve the
submission in its current form. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Xu JQ, Zhang WY, Fu JJ, Fang XZ, Gao CG,
Li C, Yao L, Li QL, Yang XB, Ren LH, et al: Viral sepsis:
Diagnosis, clinical features, pathogenesis, and clinical
considerations. Mil Med Res. 11:782024.PubMed/NCBI
|
|
2
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scheer C, Gründling M and Kuhn SO: Do not
forget the blood cultures! Intensive Care Med. 48:509–510. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dantes RB, Kaur H, Bouwkamp BA, Haass KA,
Patel P, Dudeck MA, Srinivasan A, Magill SS, Wilson WW, Whitaker M,
et al: Sepsis program activities in acute care hospitals-national
healthcare safety network, United States, 2022. MMWR Morb Mortal
Wkly Rep. 72:907–911. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weng L, Xu Y, Yin P, Wang Y, Chen Y, Liu
W, Li S, Peng JM, Dong R, Hu XY, et al: National incidence and
mortality of hospitalized sepsis in China. Crit Care. 27:842023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang R, Xu Y, Fang Y, Wang C, Xue Y, Wang
F, Cheng J, Ren H, Wang J, Guo W, et al: Pathogenetic mechanisms of
septic cardiomyopathy. J Cell Physiol. 237:49–58. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hollenberg SM and Singer M:
Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol.
18:424–434. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khatun J, Gelles JD and Chipuk JE: Dynamic
death decisions: How mitochondrial dynamics shape cellular
commitment to apoptosis and ferroptosis. Dev Cell. 59:2549–2565.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ahola S and Langer T: Ferroptosis in
mitochondrial cardiomyopathy. Trends Cell Biol. 34:150–160. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song J, Fang X, Zhou K, Bao H and Li L:
Sepsis induced cardiac dysfunction and pathogenetic mechanisms
(Review). Mol Med Rep. 28:2272023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ye H, Hu H, Zhou X, Dong M and Ren J:
Targeting ferroptosis in the maintenance of mitochondrial
homeostasis in the realm of septic cardiomyopathy. Curr Opin
Pharmacol. 74:1024302024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carbone F, Liberale L, Preda A, Schindler
TH and Montecucco F: Septic cardiomyopathy: From pathophysiology to
the clinical setting. Cells. 11:28332022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Martin L, Derwall M, Al Zoubi S,
Zechendorf E, Reuter DA, Thiemermann C and Schuerholz T: The septic
heart: Current understanding of molecular mechanisms and clinical
implications. Chest. 155:427–437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hiraiwa H, Kasugai D, Okumura T and
Murohara T: Clinical implications of septic cardiomyopathy: A
narrative review. Medicine (Baltimore). 103:e379402024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J, Wang XT, Liu DW, Zhang HM and Su
LX: Induction and deduction in sepsis-induced cardiomyopathy: Five
typical categories. Chin Med J (Engl). 133:2205–2211. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu H, Xu C, Hu Q and Wang Y:
Sepsis-induced cardiomyopathy: Understanding pathophysiology and
clinical implications. Arch Toxicol. Nov 27–2024.(Epub ahead of
print).
|
|
23
|
Fan D and Wu R: Mechanisms of the septic
heart: From inflammatory response to myocardial edema. J Mol Cell
Cardiol. 195:73–82. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lim GB: Cardiac-resident macrophages
protect against sepsis-induced cardiomyopathy. Nat Rev Cardiol.
20:1412023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hernández-Jiménez E, Plata-Menchaca EP,
Berbel D, López de Egea G, Dastis-Arias M, García-Tejada L, Sbraga
F, Malchair P, García Muñoz N, Larrad Blasco A, et al: Assessing
sepsis-induced immunosuppression to predict positive blood
cultures. Front Immunol. 15:14475232024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang H, Qin S, Li Z, Gao W, Tang M and
Dong X: Early immune system alterations in patients with septic
shock. Front Immunol. 14:11268742023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bi CF, Liu J, Yang LS and Zhang JF:
Research progress on the mechanism of sepsis induced myocardial
injury. J Inflamm Res. 15:4275–4290. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang Z, Ji S, Liu L, Liu S, Wang B, Ma Y
and Cao X: Promotion of TLR7-MyD88-dependent inflammation and
autoimmunity in mice through stem-loop changes in Lnc-Atg16l1. Nat
Commun. 15:102242024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo Q, Jin Y, Chen X, Ye X, Shen X, Lin M,
Zeng C, Zhou T and Zhang J: NF-κB in biology and targeted therapy:
New insights and translational implications. Signal Transduct
Target Ther. 9:532024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Siebeler R, de Winther MPJ and Hoeksema
MA: The regulatory landscape of macrophage interferon signaling in
inflammation. J Allergy Clin Immunol. 152:326–337. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y, Li X, Dai Y, Han Y, Wei X, Wei G,
Chen W, Kong S, He Y, Liu H, et al: Neutrophil N1 polarization
induced by cardiomyocyte-derived extracellular vesicle miR-9-5p
aggravates myocardial ischemia/reperfusion injury. J
Nanobiotechnology. 22:6322024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang L, Liu S, Wu Q, Chen R, Jiang S and
Yang Z: m6A-mediated upregulation of miRNA-193a aggravates
cardiomyocyte apoptosis and inflammatory response in sepsis-induced
cardiomyopathy via the METTL3/miRNA-193a/BCL2L2 pathway. Exp Cell
Res. 430:1137122023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Flemming A: Insights into immune
cell-fibroblast communication in heart disease. Nat Rev Immunol.
24:8492024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JGF, Colucci WS, Butler J, Voors
AA, et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stanzani G, Duchen MR and Singer M: The
role of mitochondria in sepsis-induced cardiomyopathy. Biochim
Biophys Acta Mol Basis Dis. 1865:759–773. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin Y, Xu Y and Zhang Z: Sepsis-induced
myocardial dysfunction (SIMD): The pathophysiological mechanisms
and therapeutic strategies targeting mitochondria. Inflammation.
43:1184–1200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan Y, Guan B, Xu J, Zhang H, Yi L and
Yang Z: Role of toll-like receptor-mediated pyroptosis in
sepsis-induced cardiomyopathy. Biomed Pharmacother. 167:1154932023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ni D, Lin X, Deng C, Yuan L, Li J, Liu Y,
Liang P and Jiang B: Energy metabolism: From physiological changes
to targets in sepsis-induced cardiomyopathy. Hellenic J Cardiol.
80:96–106. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu F, Zhang YT, Teng F, Li HH and Guo SB:
S100a8/a9 contributes to sepsis-induced cardiomyopathy by
activating ERK1/2-Drp1-mediated mitochondrial fission and
respiratory dysfunction. Int Immunopharmacol. 115:1097162023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vilas-Boas EA, Cabral-Costa JV, Ramos VM,
Caldeira da Silva CC and Kowaltowski AJ: Goldilocks calcium
concentrations and the regulation of oxidative phosphorylation: Too
much, too little, or just right. J Biol Chem. 299:1029042023.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun
MR, Li P, Liu B and Yang H: Songorine promotes cardiac
mitochondrial biogenesis via Nrf2 induction during sepsis. Redox
Biol. 38:1017712021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ajoolabady A, Chiong M, Lavandero S,
Klionsky DJ and Ren J: Mitophagy in cardiovascular diseases:
Molecular mechanisms, pathogenesis, and treatment. Trends Mol Med.
28:836–849. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Y, Jasper H, Toan S, Muid D, Chang X
and Zhou H: Mitophagy coordinates the mitochondrial unfolded
protein response to attenuate inflammation-mediated myocardial
injury. Redox Biol. 45:1020492021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen S, Li Q, Shi H, Li F, Duan Y and Guo
Q: New insights into the role of mitochondrial dynamics in
oxidative stress-induced diseases. Biomed Pharmacother.
178:1170842024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen A, Huang H, Fang S and Hang Q: ROS: A
‘booster’ for chronic inflammation and tumor metastasis. Biochim
Biophys Acta Rev Cancer. 1879:1891752024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu S, Huang B, Cao J, Wang Y, Xiao H, Zhu
Y and Zhang H: ROS fine-tunes the function and fate of immune
cells. Int Immunopharmacol. 119:1100692023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kuroshima T, Kawaguchi S and Okada M:
Current perspectives of mitochondria in sepsis-induced
cardiomyopathy. Int J Mol Sci. 25:47102024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sheng SY, Li JM, Hu XY and Wang Y:
Regulated cell death pathways in cardiomyopathy. Acta Pharmacol
Sin. 44:1521–1535. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jarocki M, Turek K, Saczko J, Tarek M and
Kulbacka J: Lipids associated with autophagy: Mechanisms and
therapeutic targets. Cell Death Discov. 10:4602024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Iba T, Helms J, Maier CL, Ferrer R and
Levy JH: Autophagy and autophagic cell death in sepsis: Friend or
foe? J Intensive Care. 12:412024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li J, Teng D, Jia W, Gong L, Dong H, Wang
C, Zhang L, Xu B, Wang W, Zhong L, et al: PLD2 deletion ameliorates
sepsis-induced cardiomyopathy by suppressing cardiomyocyte
pyroptosis via the NLRP3/caspase 1/GSDMD pathway. Inflamm Res.
73:1033–1046. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu X, Li Y, Zhang S and Zhou X:
Ferroptosis as a novel therapeutic target for cardiovascular
disease. Theranostics. 11:3052–3059. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang Z, Liu Y, Li Z, Feng S, Lin S, Ge Z,
Fan Y, Wang Y, Wang X and Mao J: Coronary microvascular dysfunction
and cardiovascular disease: Pathogenesis, associations and
treatment strategies. Biomed Pharmacother. 164:1150112023.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Piamsiri C, Fefelova N, Pamarthi SH,
Gwathmey JK, Chattipakorn SC, Chattipakorn N and Xie LH: Potential
roles of IP3 receptors and calcium in programmed cell death and
implications in cardiovascular diseases. Biomolecules. 14:13342024.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang J, Zhang R, Jiang X, Lv J, Li Y, Ye
H, Liu W, Wang G, Zhang C, Zheng N, et al: Toll-like receptor
4-induced ryanodine receptor 2 oxidation and sarcoplasmic reticulum
Ca2+ leakage promote cardiac contractile dysfunction in
sepsis. J Biol Chem. 293:794–807. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Carrara M, Ferrario M, Bollen Pinto B and
Herpain A: The autonomic nervous system in septic shock and its
role as a future therapeutic target: A narrative review. Ann
Intensive Care. 11:802021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang Z, Zhang D, Lin Q and Cui X:
Therapeutically fine-tuning autonomic nervous system to treat
sepsis: A new perspective on the immunomodulatory effects of
acupuncture. J Inflamm Res. 17:4373–4387. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu S and Chong W: Roles of LncRNAs in
regulating mitochondrial dysfunction in septic cardiomyopathy.
Front Immunol. 12:8020852021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sun S, Shen J, Jiang J, Wang F and Min J:
Targeting ferroptosis opens new avenues for the development of
novel therapeutics. Signal Transduct Target Ther. 8:3722023.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao
N, Sun B and Wang G: Ferroptosis: Past, present and future. Cell
Death Dis. 11:882020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen X, Yu C, Kang R, Kroemer G and Tang
D: Cellular degradation systems in ferroptosis. Cell Death Differ.
28:1135–1148. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Su Z, Liu Y, Wang L and Gu W: Regulation
of SLC7A11 as an unconventional checkpoint in tumorigenesis through
ferroptosis. Genes Dis. 12:1012542024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen J, Ma B, Yang Y, Wang B, Hao J and
Zhou X: Disulfidptosis decoded: A journey through cell death
mysteries, regulatory networks, disease paradigms and future
directions. Biomark Res. 12:452024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Niu B, Liao K, Zhou Y, Wen T, Quan G, Pan
X and Wu C: Application of glutathione depletion in cancer therapy:
Enhanced ROS-based therapy, ferroptosis, and chemotherapy.
Biomaterials. 277:1211102021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang H, Guo M, Wei H and Chen Y: Targeting
p53 pathways: Mechanisms, structures, and advances in therapy.
Signal Transduct Target Ther. 8:922023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu
J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al: AMPK-mediated BECN1
phosphorylation promotes ferroptosis by directly blocking system
Xc− activity. Curr Biol. 28:2388–2399.e5.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ichihara G, Katsumata Y, Sugiura Y,
Matsuoka Y, Maeda R, Endo J, Anzai A, Shirakawa K, Moriyama H,
Kitakata H, et al: MRP1-dependent extracellular release of
glutathione induces cardiomyocyte ferroptosis after
ischemia-reperfusion. Circ Res. 133:861–876. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen C, Xie B, Li Z, Chen L, Chen Y, Zhou
J, Ju S, Zhou Y, Zhang X, Zhuo W, et al: Fascin enhances the
vulnerability of breast cancer to erastin-induced ferroptosis. Cell
Death Dis. 13:1502022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gan B: Mitochondrial regulation of
ferroptosis. J Cell Biol. 220:e2021050432021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu Y, Wan Y, Jiang Y, Zhang L and Cheng
W: GPX4: The hub of lipid oxidation, ferroptosis, disease and
treatment. Biochim Biophys Acta Rev Cancer. 1878:1888902023.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu J, Tang D and Kang R: Targeting GPX4
in ferroptosis and cancer: Chemical strategies and challenges.
Trends Pharmacol Sci. 45:666–670. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tang Z, Li J, Peng L, Xu F, Tan Y, He X,
Zhu C, Zhang ZM, Zhang Z, Sun P, et al: Novel covalent probe
selectively targeting glutathione peroxidase 4 in vivo: Potential
applications in pancreatic cancer therapy. J Med Chem.
67:1872–1887. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Giustizieri M, Petrillo S, D'Amico J,
Torda C, Quatrana A, Vigevano F, Specchio N, Piemonte F and
Cherubini E: The ferroptosis inducer RSL3 triggers interictal
epileptiform activity in mice cortical neurons. Front Cell
Neurosci. 17:12137322023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen T, Leng J, Tan J, Zhao Y, Xie S, Zhao
S, Yan X, Zhu L, Luo J, Kong L and Yin Y: Discovery of novel potent
covalent glutathione peroxidase 4 inhibitors as highly selective
ferroptosis inducers for the treatment of triple-negative breast
cancer. J Med Chem. 66:10036–10059. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang Y, Swanda RV, Nie L, Liu X, Wang C,
Lee H, Lei G, Mao C, Koppula P, Cheng W, et al: mTORC1 couples
cyst(e)ine availability with GPX4 protein synthesis and ferroptosis
regulation. Nat Commun. 12:15892021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ru Q, Li Y, Chen L, Wu Y, Min J and Wang
F: Iron homeostasis and ferroptosis in human diseases: Mechanisms
and therapeutic prospects. Signal Transduct Target Ther. 9:2712024.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Grange C, Lux F, Brichart T, David L,
Couturier A, Leaf DE, Allaouchiche B and Tillement O: Iron as an
emerging therapeutic target in critically ill patients. Crit Care.
27:4752023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Roemhild K, von Maltzahn F, Weiskirchen R,
Knüchel R, von Stillfried S and Lammers T: Iron metabolism:
Pathophysiology and pharmacology. Trends Pharmacol Sci. 42:640–656.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Salnikow K: Role of iron in cancer. Semin
Cancer Biol. 76:189–194. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zou Y, Yang A, Chen B, Deng X, Xie J, Dai
D, Zhang J, Tang H, Wu T, Zhou Z, et al: crVDAC3 alleviates
ferroptosis by impeding HSPB1 ubiquitination and confers
trastuzumab deruxtecan resistance in HER2-low breast cancer. Drug
Resist Updat. 77:1011262024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wang F, Wang J, Shen Y, Li H, Rausch WD
and Huang X: Iron dyshomeostasis and ferroptosis: A new alzheimer's
disease hypothesis? Front Aging Neurosci. 14:8305692022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen F, Kang R, Tang D and Liu J:
Ferroptosis: Principles and significance in health and disease. J
Hematol Oncol. 17:412024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Gasmi A, Bjørklund G, Mujawdiya PK,
Semenova Y, Piscopo S and Peana M: Coenzyme Q10 in aging
and disease. Crit Rev Food Sci Nutr. 64:3907–3919. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Nakamura T, Hipp C, Santos Dias Mourão A,
Borggräfe J, Aldrovandi M, Henkelmann B, Wanninger J, Mishima E,
Lytton E, Emler D, et al: Phase separation of FSP1 promotes
ferroptosis. Nature. 619:371–377. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Roh JL: Targeting ferroptosis suppressor
protein 1 in cancer therapy: Implications and perspectives, with
emphasis on head and neck cancer. Crit Rev Oncol Hematol.
202:1044402024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu Y, Lu S, Wu LL, Yang L, Yang L and
Wang J: The diversified role of mitochondria in ferroptosis in
cancer. Cell Death Dis. 14:5192023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mishima E, Nakamura T, Zheng J, Zhang W,
Mourão ASD, Sennhenn P and Conrad M: DHODH inhibitors sensitize to
ferroptosis by FSP1 inhibition. Nature. 619:E9–E18. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Liang D, Feng Y, Zandkarimi F, Wang H,
Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR and Jiang X: Ferroptosis
surveillance independent of GPX4 and differentially regulated by
sex hormones. Cell. 186:2748–2764.e22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
No authors listed. Sex hormone signaling
suppresses ferroptosis via phospholipid remodeling. Cancer Discov.
13:17592023. View Article : Google Scholar
|
|
96
|
Nakamura T and Conrad M: Exploiting
ferroptosis vulnerabilities in cancer. Nat Cell Biol. 26:1407–1419.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang W, Dai J, Hou G, Liu H, Zheng S,
Wang X, Lin Q, Zhang Y, Lu M, Gong Y, et al: SMURF2 predisposes
cancer cell toward ferroptosis in GPX4-independent manners by
promoting GSTP1 degradation. Mol Cell. 83:4352–4369.e8. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Micangeli G, Menghi M, Profeta G, Tarani
F, Mariani A, Petrella C, Barbato C, Ferraguti G, Ceccanti M,
Tarani L and Fiore M: The impact of oxidative stress on pediatrics
syndromes. Antioxidants (Basel). 11:19832022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Cheng X, Yu C, Yang X, Wang F and Min J: A
panoramic view of ferroptosis in cardiovascular disease. Kidney Dis
(Basel). 9:173–186. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu G, Xie X, Liao W, Chen S, Zhong R, Qin
J, He P and Xie J: Ferroptosis in cardiovascular disease. Biomed
Pharmacother. 170:1160572024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Fang W, Xie S and Deng W: Ferroptosis
mechanisms and regulations in cardiovascular diseases in the past,
present, and future. Cell Biol Toxicol. 40:172024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Liu BH, Xu CZ, Liu Y, Lu ZL, Fu TL, Li GR,
Deng Y, Luo GQ, Ding S, Li N and Geng Q: Mitochondrial quality
control in human health and disease. Mil Med Res.
11:322024.PubMed/NCBI
|
|
104
|
Long X, Liu M, Nan Y, Chen Q, Xiao Z,
Xiang Y, Ying X, Sun J, Huang Q and Ai K: Revitalizing ancient
mitochondria with nano-strategies: Mitochondria-remedying nanodrugs
concentrate on disease control. Adv Mater. 36:e23082392024.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Conrad M and Proneth B: Broken hearts:
Iron overload, ferroptosis and cardiomyopathy. Cell Res.
29:263–264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Liu C, Zou Q, Tang H, Liu J, Zhang S, Fan
C, Zhang J, Liu R, Liu Y, Liu R, et al: Melanin nanoparticles
alleviate sepsis-induced myocardial injury by suppressing
ferroptosis and inflammation. Bioact Mater. 24:313–321.
2022.PubMed/NCBI
|
|
107
|
Liu R, Li F, Hao S, Hou D, Zeng X, Huang
H, Sethi G, Guo J and Duan C: Low-dose olaparib improves septic
cardiac function by reducing ferroptosis via accelerated mitophagy
flux. Pharmacol Res. 200:1070562024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Dan Z, Shi X, Shu C, Zhu R, Wang Y and Zhu
H: 4-amino-2-trifluoromethyl-phenyl retinate alleviates
lipopolysaccharide-induced acute myocardial injury through
activation of the KLF4/p62 axis. Cell Signal. 114:1110012024.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kanwar P, Samtani H, Sanyal SK, Srivastava
AK, Suprasanna P and Pandey GK: VDAC and its interacting partners
in plant and animal systems: An overview. Crit Rev Biotechnol.
40:715–732. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
She H, Tan L, Du Y, Zhou Y, Guo N, Zhang
J, Du Y, Wang Y, Wu Z, Ma C, et al: VDAC2 malonylation participates
in sepsis-induced myocardial dysfunction via mitochondrial-related
ferroptosis. Int J Biol Sci. 19:3143–3158. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yu H, Liu J, Bu X, Ma Z, Yao Y, Li J,
Zhang T, Song W, Xiao X, Sun Y, et al: Targeting METTL3 reprograms
the tumor microenvironment to improve cancer immunotherapy. Cell
Chem Biol. 31:776–791.e7. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Shen H, Xie K, Tian Y and Wang X:
N6-methyladenosine writer METTL3 accelerates the sepsis-induced
myocardial injury by regulating m6A-dependent ferroptosis.
Apoptosis. 28:514–524. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Cao G, Zeng Y, Zhao Y, Lin L, Luo X, Guo
L, Zhang Y and Cheng Q: H2S regulation of ferroptosis attenuates
sepsis-induced cardiomyopathy. Mol Med Rep. 26:3352022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Guo L, Li P, Wang Y, Wang J, Lei J, Zhao
J, Wu X, He W, Jia J, Miao J, et al: Yiqifumai injection
ameliorated sepsis-induced cardiomyopathy by inhibition of
ferroptosis via XCT/GPX4 axis. Shock. 61:638–645. 2024.PubMed/NCBI
|
|
115
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mukherjee R, Tetri LH, Li SJ, Fajardo G,
Ostberg NP, Tsegay KB, Gera K, Cornell TT, Bernstein D,
Mochly-Rosen D and Haileselassie B: Drp1/p53 interaction mediates
p53 mitochondrial localization and dysfunction in septic
cardiomyopathy. J Mol Cell Cardiol. 177:28–37. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gao N, Tang AL, Liu XY, Chen J and Zhang
GQ: p53-dependent ferroptosis pathways in sepsis. Int
Immunopharmacol. 118:1100832023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Lin X, Zhao X, Chen Q, Wang X, Wu Y and
Zhao H: Quercetin ameliorates ferroptosis of rat cardiomyocytes via
activation of the SIRT1/p53/SLC7A11 signaling pathway to alleviate
sepsis-induced cardiomyopathy. Int J Mol Med. 52:1162023.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Qin S, Ren Y, Feng B, Wang X, Liu J, Zheng
J, Li K, Chen M, Chen T, Mei H and Fu X: ANXA1sp protects against
sepsis-induced myocardial injury by inhibiting ferroptosis-induced
cardiomyocyte death via SIRT3-mediated p53 deacetylation. Mediators
Inflamm. 2023:66389292023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mishima E and Conrad M: Nutritional and
metabolic control of ferroptosis. Annu Rev Nutr. 42:275–309. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhu M, Peng L, Huo S, Peng D, Gou J, Shi
W, Tao J, Jiang T, Jiang Y, Wang Q, et al: STAT3 signaling promotes
cardiac injury by upregulating NCOA4-mediated ferritinophagy and
ferroptosis in high-fat-diet fed mice. Free Radic Biol Med.
201:111–125. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wang Y, Ding H, Zheng Y, Wei X, Yang X,
Wei H, Tian Y, Sun X, Wei W, Ma J, et al: Alleviated NCOA4-mediated
ferritinophagy protected RA FLSs from ferroptosis in
lipopolysaccharide-induced inflammation under hypoxia. Inflamm Res.
73:363–379. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Babaei-Abraki S, Karamali F and
Nasr-Esfahani MH: Ferroptosis: The functions of Nrf2 in human
embryonic stem cells. Cell Signal. 106:1106542023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Chang LC, Chiang SK, Chen SE, Yu YL, Chou
RH and Chang WC: Heme oxygenase-1 mediates BAY 11-7085 induced
ferroptosis. Cancer Lett. 416:124–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Dodson M, Castro-Portuguez R and Zhang DD:
NRF2 plays a critical role in mitigating lipid peroxidation and
ferroptosis. Redox Biol. 23:1011072019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Pietrangelo A: Ferroportin disease:
Pathogenesis, diagnosis and treatment. Haematologica.
102:1972–1984. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang J, Guan P, Chen Y, Xu M, Wang N and
Ji E: Cyclovirobuxine D pretreatment ameliorates septic heart
injury through mitigation of ferroptosis. Exp Ther Med. 26:4072023.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kong C, Ni X, Wang Y, Zhang A, Zhang Y,
Lin F, Li S, Lv Y, Zhu J, Yao X, et al: ICA69 aggravates
ferroptosis causing septic cardiac dysfunction via STING
trafficking. Cell Death Discov. 8:1872022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Gu Q, Xu F, Orgil BO, Khuchua Z,
Munkhsaikhan U, Johnson JN, Alberson NR, Pierre JF, Black DD, Dong
D, et al: Systems genetics analysis defines importance of
TMEM43/LUMA for cardiac- and metabolic-related pathways. Physiol
Genomics. 54:22–35. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Chen Z, Cao Z, Gui F, Zhang M, Wu X, Peng
H, Yu B, Li W, Ai F and Zhang J: TMEM43 protects against
sepsis-induced cardiac injury via inhibiting ferroptosis in mice.
Cells. 11:29922022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Deng W, Ren G, Luo J, Gao S, Huang W, Liu
W and Ye S: TRPM7 mediates endoplasmic reticulum stress and
ferroptosis in sepsis-induced myocardial injury. J Bioenerg
Biomembr. 55:207–217. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Jiang C, Shi Q, Yang J, Ren H, Zhang L,
Chen S, Si J, Liu Y, Sha D, Xu B and Ni J: Ceria nanozyme
coordination with curcumin for treatment of sepsis-induced cardiac
injury by inhibiting ferroptosis and inflammation. J Adv Res.
63:159–170. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jiao Y, Zhang Q, Zhang J, Zha Y, Wang J,
Li Y and Zhang S: Platelet-rich plasma ameliorates
lipopolysaccharide-induced cardiac injury by inflammation and
ferroptosis regulation. Front Pharmacol. 13:10266412022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhou B, Zhang J, Chen Y, Liu Y, Tang X,
Xia P, Yu P and Yu S: Puerarin protects against sepsis-induced
myocardial injury through AMPK-mediated ferroptosis signaling.
Aging (Albany NY). 14:3617–3632. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Xiao Y, Yu Y, Hu L, Yang Y, Yuan Y, Zhang
W, Luo J and Yu L: Correction to: Matrine alleviates sepsis-induced
myocardial injury by inhibiting ferroptosis and apoptosis.
Inflammation. 47:15452024. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Fang X, Fu W, Zou B and Zhang F:
Tectorigenin relieved sepsis-induced myocardial ferroptosis by
inhibiting the expression of Smad3. Toxicol Res (Camb). 12:520–526.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zeng Y, Cao G, Lin L, Zhang Y, Luo X, Ma
X, Aiyisake A and Cheng Q: Resveratrol attenuates sepsis-induced
cardiomyopathy in rats through anti-ferroptosis via the Sirt1/Nrf2
pathway. J Invest Surg. 36:21575212023. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Wang X, Simayi A, Fu J, Zhao X and Xu G:
Resveratrol mediates the miR-149/HMGB1 axis and regulates the
ferroptosis pathway to protect myocardium in endotoxemia mice. Am J
Physiol Endocrinol Metab. 323:E21–E32. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Gao J, Luo T and Wang J: Gene
interfered-ferroptosis therapy for cancers. Nat Commun.
12:53112021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Vinik Y, Maimon A, Dubey V, Raj H,
Abramovitch I, Malitsky S, Itkin M, Ma'ayan A, Westermann F,
Gottlieb E, et al: Programming a ferroptosis-to-apoptosis
transition landscape revealed ferroptosis biomarkers and repressors
for cancer therapy. Adv Sci (Weinh). 11:e23072632024. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Huang Y, Li L, Li Y, Lu N, Qin H, Wang R,
Li W, Cheng Z, Li Z, Kang P, et al: Knockdown of LncRNA Lcn2-204
alleviates sepsis-induced myocardial injury by regulation of iron
overload and ferroptosis. J Mol Cell Cardiol. 192:79–93. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Song J, Ren K, Zhang D, Lv X, Sun L, Deng
Y and Zhu H: A novel signature combing cuproptosis- and
ferroptosis-related genes in sepsis-induced cardiomyopathy. Front
Genet. 14:11707372023. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zou HX, Hu T, Zhao JY, Qiu BQ, Zou CC, Xu
QR, Liu JC, Lai SQ and Huang H: Exploring dysregulated
ferroptosis-related genes in septic myocardial injury based on
human heart transcriptomes: Evidence and new insights. J Inflamm
Res. 16:995–1015. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lin H, Ji F, Lin KQ, Zhu YT, Yang W, Zhang
LH, Zhao JG and Pei YH: LPS-aggravated ferroptosis via disrupting
circadian rhythm by Bmal1/AKT/p53 in sepsis-induced myocardial
injury. Inflammation. 46:1133–1143. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Xu Y and Bu G: Identification of two novel
ferroptosis-associated targets in sepsis-induced cardiac injury:
Hmox1 and Slc7a11. Front Cardiovasc Med. 10:11859242023. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lu SM, Yang B, Tan ZB, Wang HJ, Xie JD,
Xie MT, Jiang WH, Huang JZ, Li J, Zhang L, et al: TaoHe ChengQi
decoction ameliorates sepsis-induced cardiac dysfunction through
anti-ferroptosis via the Nrf2 pathway. Phytomedicine.
129:1555972024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Lu JS, Wang JH, Han K and Li N: Nicorandil
regulates ferroptosis and mitigates septic cardiomyopathy via
TLR4/SLC7A11 signaling pathway. Inflammation. 47:975–988. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Singh D, Singh R and Akindele AJ:
Therapeutic potential of nicorandil beyond anti-anginal drug: A
review on current and future perspectives. Heliyon. 10:e289222024.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Zeng T, Zhou Y, Yu Y, Wang JW, Wu Y, Wang
X, Zhu L, Zhou LM and Wan LH: rmMANF prevents sepsis-associated
lung injury via inhibiting endoplasmic reticulum stress-induced
ferroptosis in mice. Int Immunopharmacol. 114:1096082023.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Jiang W, Ren J, Zhou H, He R, Li D, Xiong
R, He Z and Cheng D: TMEM16A deficiency in alveolar type 2
epithelial cells protected against endoplasmic reticulum
stress-induced ferroptosis during acute lung injury. Int
Immunopharmacol. 125:1112082023. View Article : Google Scholar : PubMed/NCBI
|