Introduction
Reactive oxygen species (ROS) are highly reactive
oxygen free radicals or non-radical molecules, which include
hydrogen peroxide (H2O2), superoxide anion
(O2•−) and hydroxyl radical (•OH)
(1). These molecules regulate a
number of cellular events, including transcription factor
activation, gene expression, differentiation and cell proliferation
(2,3). ROS are mainly formed as by-products of
the respiratory chain during oxidative phosphorylation in the form
of O2•− or are specifically produced by
oxidases, such as nicotine adenine diphosphate (NADPH) oxidase,
xanthine oxidase and arachidonic acid oxygenases (4). Excessive ROS production induces
cellular damage and death (5,6).
Therefore, there are various antioxidants and systems to control
excessive ROS levels.
Thioredoxin (Trx) is a low molecular weight (10- to
12-kDa) redox protein (7), which
affects cell growth and proliferation by regulating the redox
status in cells (8). Trx has two
main isoforms: The cytosolic form, Trx-1, and the mitochondrial
form, Trx-2 (9). These Trxs are
reduced back by Trx reductase and NADPH following the reduction of
oxidative target proteins (10,11).
It has been reported that Trx-1 is implicated in cell survival,
tumor development, angiogenesis and chemoresistance (12,13).
Numerous studies have demonstrated that the overexpression of Trx
occurs in a variety of cancer types, including gastric and lung
cancers (8,14). PX-12 (1-methylpropyl 2-imidazolyl
disulfide) is an irreversible Trx-1 inhibitor, which has antitumor
properties (15). PX-12 decreased
the activity of Trx-1 by thioalkylating the critical cysteine
residue (Cys73) in this protein or by increasing the dimerization
of its oxidative form. It has also been reported that PX-12
decreases hypoxia-inducible factor-1α transactivation and vascular
endothelial growth factor (16,17).
Therefore, PX-12 has been clinically tested in colorectal, lung and
pancreatic cancers (18,19).
Cervical cancer is a major cause of mortality in
females worldwide. Its carcinogenesis is associated with excessive
inflammation mediated by ROS. An increase in Trx-1 levels has been
observed in cervical cancer patients compared with a control group
(20). However, little is known
about the cellular effect of PX-12 in cervical cancer.
PX-12-induced cell death in cervical cancer cells may be
toxicologically attractive in relation to intracellular ROS levels.
Therefore, in the present study, the effects of PX-12 on cell
growth and death were investigated in human cervical adenocarcinoma
HeLa cells. The effects of various caspase inhibitors (pan-caspase
and caspase-3, -8 and -9), N-acetyl cysteine (NAC; a well known
antioxidant) and L-buthionine sulfoximine [BSO; an inhibitor of
glutathione (GSH) synthesis] were also evaluated in PX-12-treated
HeLa cells with respect to cell growth, cell death and ROS and GSH
levels.
Materials and methods
Cell culture
Human cervical adenocarcinoma HeLa cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and maintained in a humidified incubator containing 5%
CO2 at 37°C. The HeLa cells were cultured in RPMI-1640
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (Sigma-Aldrich) and 1% penicillin-streptomycin (Gibco
BRL, Grand Island, NY, USA). The cells were routinely grown in
100-mm plastic tissue culture dishes (Nunc, Roskilde, Denmark) and
harvested with a solution of trypsin-EDTA while in a logarithmic
phase of growth.
Reagents
PX-12 was purchased from Tocris Bioscience (Bristol,
UK) and was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich)
at 100 mM as a stock solution. The pan-caspase inhibitor
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK),
caspase-3 inhibitor
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone (Z-DEVD-FMK),
caspase-8 inhibitor
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone (Z-IETD-FMK)
and caspase-9 inhibitor
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone (Z-LEHD-FMK)
were obtained from R&D Systems Inc. (Minneapolis, MN, USA) and
were dissolved in DMSO at 10 mM to serve as stock solutions. NAC
and BSO were obtained from Sigma-Aldrich. NAC was dissolved in 20
mM HEPES buffer (pH 7.0) and BSO was dissolved in water. Based on
previous studies (21,22), cells were pretreated with 15 μM
caspase inhibitors, 2 mM NAC or 10 μM BSO for 1 h prior to
treatment with PX-12. DMSO (0.2%) was used as a control vehicle and
it did not affect cell growth or death.
Growth inhibition assay
The effect of PX-12 on cell growth was determined by
measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Sigma-Aldrich) absorbance in living cells as
described previously (23). In
brief, 1×104 cells/well were seeded in 96-well
microtiter plates (Nunc). Following exposure to the designated
doses of PX-12 for the indicated times, MTT solution [20 μl: 2
mg/ml in phosphate-buffered saline (PBS)] was added to each well.
The plates were incubated for 3 h at 37°C. Medium was withdrawn
from the plates by pipetting and 200 μl DMSO was added to each well
to solubilize the formazan crystals. The optical density was
measured at 570 nm using a microplate reader (Synergy™ 2, BioTek
Instruments Inc., Winooski, VT, USA). The cell population was
visualized under a light microscope at ×400 magnification
(FLoid® Cell Imaging Station, Life Technologies
Corporation, Carlsbad, CA, USA).
Cell cycle and sub-G1 cell analysis
Cell cycle and sub-G1 cell analysis were determined
by propidium iodide (PI, Ex/Em=488/617 nm; Sigma-Aldrich) staining
as described previously (24). In
brief, 1×106 cells in a 60-mm culture dish (Nunc) were
incubated with the designated doses of PX-12 for 72 h. Total cells,
including floating cells, were then washed with PBS and fixed in
70% (v/v) ethanol. Cells were washed again with PBS, then incubated
with PI (10 μg/ml) with simultaneous RNase treatment at 37°C for 30
min. Cellular DNA content was measured using a FACStar flow
cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed
using Lysis II and CellFit software (Becton Dickinson).
Annexin V-fluorescein isothiocyanate
(FITC)/PI staining for the detection of cell death
Apoptotic cell death was determined by staining
cells with annexin V-FITC (Ex/Em=488/519 nm; Invitrogen Life
Technologies, Camarillo, CA, USA) as described previously (25). In brief, 1×106 cells in a
60-mm culture dish were incubated with the designated doses of
PX-12 for 72 h with or without 15 μM each caspase inhibitor, 2 mM
NAC or 10 μM BSO. Cells were washed twice with cold PBS and then
resuspended in 500 μl binding buffer [10 mM HEPES/NaOH (pH 7.4),
140 mM NaCl and 2.5 mM CaCl2] at a concentration of
1×106 cells/ml. Annexin V-FITC (5 μl) and PI (1 μg/ml)
were then added and the cells were analyzed with the FACStar flow
cytometer. Viable cells were negative for PI and annexin V,
apoptotic cells were positive for annexin V and negative for PI,
whereas late apoptotic dead cells exhibited high annexin V and PI
labeling. Non-viable cells that underwent necrosis, were positive
for PI and negative for annexin V.
Measurement of the mitochondrial membrane
potential (MMP)
MMP was measured by a rhodamine 123 fluorescent dye
(Ex/Em=485/535 nm; Sigma-Aldrich) as described previously (25,26).
In brief, 1×106 cells in a 60-mm culture dish were
incubated with the designated doses of PX-12 for 72 h with or
without 15 μM each caspase inhibitor, 2 mM NAC or 10 μM BSO. Cells
were washed twice with PBS and incubated with rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity was
determined using the FACStar flow cytometer. The cells that were
rhodamine 123-negative were indicated to have lost MMP.
Detection of intracellular ROS
levels
Intracellular ROS levels were detected using an
oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Ex/Em=495/529 nm; Invitrogen Life Technologies) and dihydroethidium
(DHE; Ex/Em=518/605 nm; Invitrogen Life Technologies) as previously
described (25,27). DHE is highly selective for
O2•− among ROS. In brief, 1×106
cells in a 60-mm culture dish were incubated with the designated
doses of PX-12 for 72 h with or without 15 μM each caspase
inhibitor, 2 mM NAC or 10 μM BSO. Cells were then washed in PBS and
incubated with 20 μM H2DCFDA or DHE at 37°C for 30 min.
H2DCFDA or DHE fluorescence was assessed using the
FACStar flow cytometer. ROS and O2•− levels
were expressed as mean fluorescence intensity, which was calculated
by CellQuest software (Becton Dickinson).
Detection of intracellular GSH
Intracellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate (CMFDA) dye (Ex/Em=522/595 nm;
Invitrogen Life Technologies) as previously described (27,28).
In brief, 1×106 cells in a 60-mm culture dish were
incubated with the designated doses of PX-12 for 72 h with or
without 15 μM each caspase inhibitor, 2 mM NAC or 10 μM BSO. Cells
were then washed with PBS and incubated with 5 μM CMFDA at 37°C for
30 min. CMFDA fluorescence intensity was determined using the
FACStar flow cytometer. Negative CMFDA staining (GSH-depletion) of
cells was expressed as the percentage of CMFDA-negative cells.
Statistical analysis
Results represent the mean of at least three
independent experiments (mean ± standard deviation). Data were
analyzed using Instat software (GraphPad Prism 4, San Diego, CA,
USA). Student’s t-test or one-way analysis of variance with post
hoc analysis using Tukey’s multiple comparison test were used for
parametric data. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of PX-12 on cell growth and cell
cycle distribution in HeLa cells
We first examined the effect of PX-12 on the growth
of HeLa cells. After exposure to 1–10 μM PX-12 for 72 h, the
population of HeLa cells was not affected at 1 μM PX-12, whereas
the population of these cells was markedly decreased at 5–10 μM
PX-12 (Fig. 1A). In addition, 5 and
10 μM PX-12 treatment induced cell death in HeLa cells (Fig. 1A). Based on MTT assays, the tested
doses (1–30 μM) of PX-12 did not affect changes in cell growth at
24 h, whereas a high dose of 30 μM PX-12 significantly decreased
the growth of HeLa cell at 48 h (Fig.
1B). At 72 h, 5–30 μM PX-12 significantly inhibited the growth
of HeLa cells with an IC50 value (the half maximal
inhibitory concentration) of ~7 μM at 72 h (Fig. 1B). When the cell cycle distributions
were examined in PX-12-treated HeLa cells, 5 and 10 μM PX-12
significantly induced a G2/M phase arrest of the cell cycle at 72 h
(Fig. 1C).
Effects of PX-12 on cell death and MMP in
HeLa cells
As shown in Fig. 2A,
PX-12 increased the percentages of sub-G1 cells in a dose-dependent
manner at 72 h. Treatment with 5–30 μM PX-12 increased the number
of annexin V-FITC-positive cells, whereas 1 μM PX-12 did not
increase the percentage of annexin V-FITC-positive cells (Fig. 2B). Cell death is closely associated
with the collapse of MMP (29). As
expected, the loss of MMP was observed in PX-12-treated HeLa cells
(Fig. 2C). This result indicates
that PX-12 damaged the membrane of mitochondria in HeLa cells.
Effects of PX-12 on ROS and GSH levels in
HeLa cells
To assess the intracellular ROS levels in
PX-12-treated HeLa cells, we used H2DCFDA and DHE dyes.
As shown in Fig. 3A, PX-12
significantly increased the intracellular ROS (H2DCFDA)
levels in HeLa cells at 72 h. Among the tested concentrations, 10
μM PX-12 led to the maximum level of ROS (H2DCFDA)
(Fig. 3A). Moreover, red
fluorescence derived from DHE reflecting the intracellular
O2•− levels was markedly increased in
PX-12-treated HeLa cells at 72 h (Fig.
3B). When intracellular GSH levels were measured in
PX-12-treated HeLa cells using a CMFDA dye, 10–30 μM PX-12
significantly increased the number of GSH-depleted cells at 72 h;
however, 5 μM PX-12 marginally induced GSH depletion (Fig. 3C).
Effects of caspase inhibitors on cell
death, MMP, O2•− and GSH levels in
PX-12-treated HeLa cells
We determined which caspases were involved in HeLa
cell death caused by PX-12. For this experiment, we selected 10 μM
PX-12 as a suitable dose to differentiate the levels of cell death
in the presence or absence of each caspase inhibitor. Based on a
previous study (21), HeLa cells
were pretreated with 15 μM caspase inhibitor for 1 h prior to
treatment with PX-12. This dose did not significantly affect cell
death in the control HeLa cells (data not shown). Treatment with
all the tested caspase inhibitors (Z-VAD for pan-caspases, Z-DEVD
for caspase-3, Z-IETD for caspase-8 and Z-LEHD for caspase-9)
demonstrated the significant rescue of HeLa cells from
PX-12-induced apoptosis at 72 h, as measured by the population of
annexin V-FITC-positive cells (Fig.
4A). In addition, all the caspase inhibitors marginally, but
not significantly, prevented the loss of MMP caused by PX-12
(Fig. 4B).
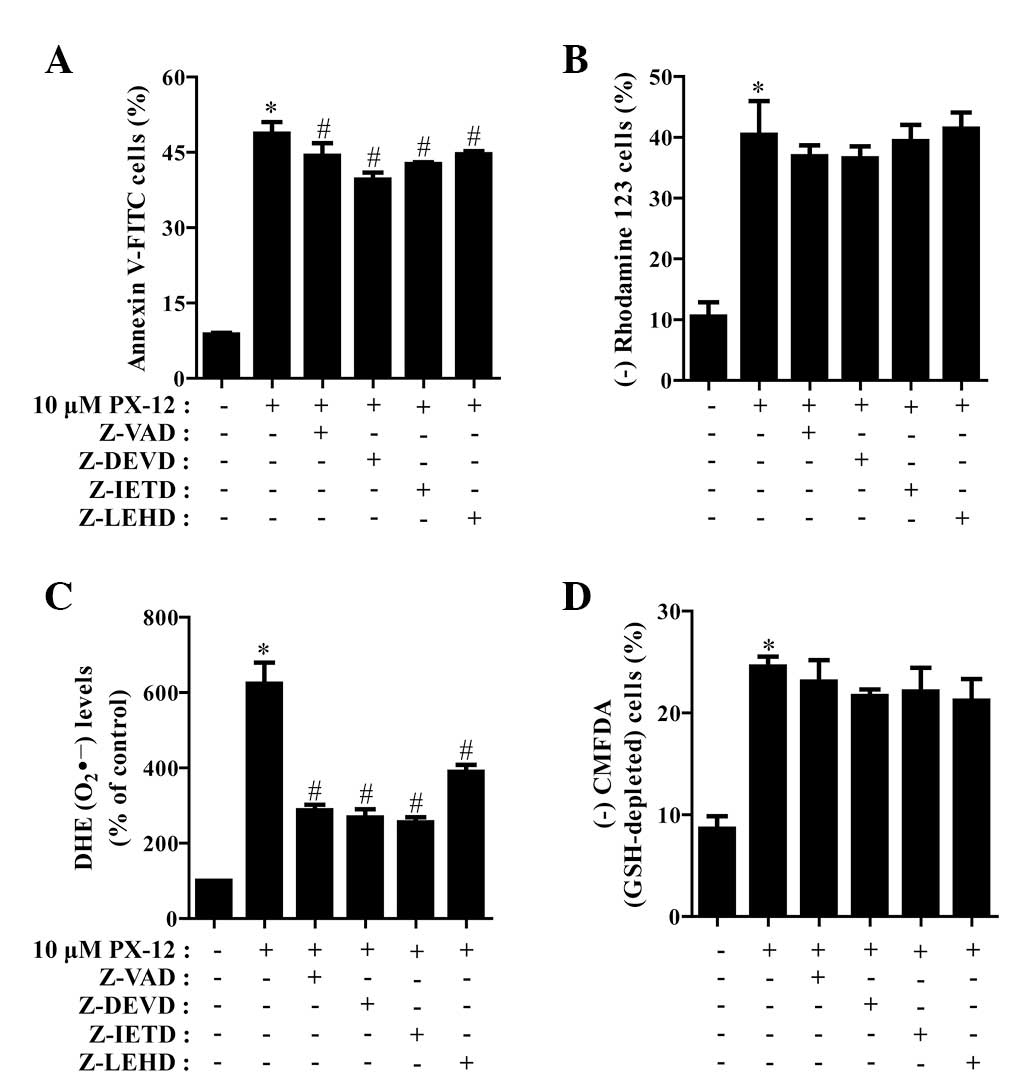 | Figure 4Effects of caspase inhibitors on cell
death, MMP, O2•− and GSH levels in
PX-12-treated HeLa cells. Exponentially growing cells were treated
with 10 μM PX-12 for 72 h following 1 h pre-incubation with 15 μM
each caspase inhibitor. The number of annexin V/PI-stained cells,
as well as MMP, O2•− and GSH levels in HeLa
cells were measured using a FACStar flow cytometer. Percentage of
(A) annexin V-positive cells, (B) rhodamine 123-negative (loss of
MMP) cells, (C) O2•− (DHE) levels and (D)
CMFDA-negative (GSH-depleted) cells. *P<0.05,
compared with the control group; #P<0.05, compared
with cells treated with PX-12 only. MMP, mitochondrial membrane
potential; GSH, glutathione; PI, propidium iodide; DHE,
dihydroethidium; CMFDA, 5-chloromethylfluorescein diacetate. |
It was also investigated whether the levels of
intracellular O2•− and GSH in PX-12-treated
HeLa cells were affected by treatment with each caspase inhibitor.
As shown in Fig. 4C, all the
caspase inhibitors significantly decreased
O2•− levels in PX-12-treated HeLa cells.
Moreover, these caspase inhibitors marginally prevented GSH
depletion in these cells (Fig.
4D).
Effects of NAC and BSO on cell death,
MMP, O2•− and GSH levels in PX-12-treated
HeLa cells
The effects of NAC or BSO on cell death and MMP in
10 μM PX-12-treated HeLa cells were assessed at 72 h. As shown in
Fig. 5A and B, NAC significantly
decreased the number of annexin V-FITC-positive cells in the
PX-12-treated HeLa cell population, whereas BSO increased the
number of these cells. NAC and BSO did not significantly affect
cell growth and cell death in the control HeLa cells (data not
shown). With respect to MMP, NAC significantly attenuated the loss
of MMP caused by PX-12 whereas BSO enhanced, to a certain extent,
the loss in these cells (Fig. 5C).
Furthermore, it was determined whether the levels of intracellular
O2•− and GSH in PX-12-treated HeLa cells were
affected by treatment with NAC or BSO. While NAC markedly decreased
the level of O2•− in PX-12-treated HeLa
cells, BSO had no effect on the level of O2•−
in these cells (Fig. 5D). With
regard to GSH levels, NAC markedly prevented GSH depletion caused
by PX-12, whereas BSO intensified GSH depletion in these cells
(Fig. 5E).
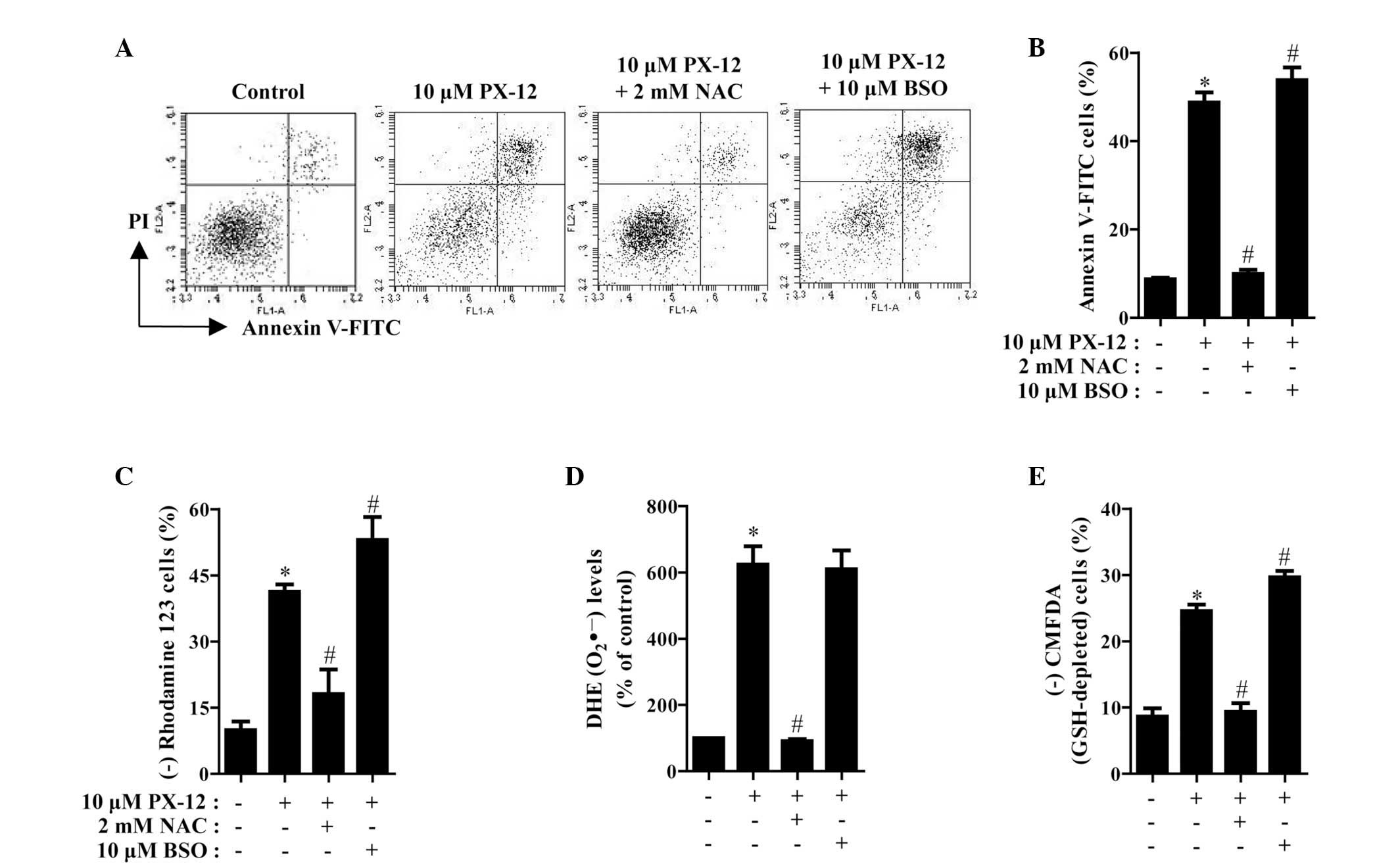 | Figure 5Effects of NAC and BSO on cell death,
MMP, O2•− and GSH levels in PX-12-treated
HeLa cells. Exponentially growing cells were treated with 10 μM
PX-12 for 72 h following 1 h pre-incubation with 2 mM NAC or 10 μM
BSO. The number of annexin V/PI-stained cells, as well as MMP,
O2•− and GSH levels in HeLa cells were
measured using a FACStar flow cytometer. (A) Each figure shows a
representative for annexin V and/or PI stained cells. (B) The graph
shows the percentage of annexin V-positive cells from A. (C) The
graph shows the percentage of rhodamine 123-negative (loss of MMP)
cells. (D and E) The graphs indicate O2•−
(DHE) levels as a percentage of the control (D) and the percentage
of CMFDA-negative (GSH-depleted) cells (E). *P<0.05,
compared with the control group; #P<0.05, compared
with cells treated with PX-12 only. NAC, N-acetyl cysteine; BSO,
L-buthionine sulfoximine; MMP, mitochondrial membrane potential;
GSH, glutathione; PI, propidium iodide; DHE, dihydroethidium;
CMFDA, 5-chloromethylfluorescein diacetate. |
Discussion
The aim of the present study was to assess the
effects of PX-12 on cell growth and death in HeLa cells in
association with ROS and GSH levels. Following exposure to PX-12
for 72 h, the IC50 value in HeLa cells was ~7 μM based
on MTT assays. However, the tested doses of PX-12 did not show the
growth inhibition of HeLa cells at 24 h and this effect was mild at
48 h. Therefore, the susceptibility of HeLa cells to PX-12 appeared
to significantly increase after the incubation time of 48 h. DNA
flow cytometric analysis indicated that 5 and 10 μM significantly
induced a G2/M phase arrest of the cell cycle. As 20 and 30 μM
PX-12 completely decreased cell growth, it was not possible to
perform cell cycle analysis in HeLa cells. Similarly, PX-12 induced
a G2/M phase arrest in B-cell lymphoma and breast cancer cells
(7,30). We also observed that PX-12 induced a
G2/M phase arrest in A549 and Calu-6 lung cancer cells (unpublished
data). Therefore, the G2/M phase arrest in PX-12-treated cells was
an underlying mechanism to suppress the growth of cancer cells,
including HeLa cells.
PX-12 also increased the number of dead cells and
annexin V-FITC-positive cells at 72 h, suggesting that
PX-12-induced HeLa cell death occurred via apoptosis. Apoptosis is
closely associated with the collapse of MMP (31). Our results demonstrated that PX-12
triggered the loss of MMP in HeLa cells in a dose-dependent manner.
Furthermore, treatment with the caspase inhibitors investigated in
this experiment significantly prevented HeLa cell death caused by
PX-12. In particular, the caspase-8 inhibitor attenuated HeLa cell
death. These data suggest that the mitochondrial pathway and cell
death receptor pathway are together necessary for the complete
induction of apoptosis in PX-12-treated HeLa cells. However, all
the caspase inhibitors marginally, but not significantly prevented
the loss of MMP caused by PX-12. These results implied that the
loss of MMP by PX-12 may not be enough to fully induce apoptosis in
HeLa cells under the inhibition of caspases by their
inhibitors.
PX-12, as an inhibitor of Trx-1, increases ROS
levels. It has been reported that PX-12 induces oxidative stress
(32). Similarly, in the present
study, the intracellular ROS levels, particularly those of
O2•−, were significantly increased in
PX-12-treated HeLa cells at 72 h. All caspase inhibitors
demonstrating anti-apoptotic effects decreased the level of
O2•−. These data indicated that the level of
O2•−, among other ROS, is closely associated
with apoptosis in PX-12-treated HeLa cells. Furthermore, NAC
markedly prevented apoptotic cell death and the loss of MMP in
PX-12-treated HeLa cells, accompanied by strongly decreasing
O2•− levels in these cells. Overall, these
results suggest that PX-12-induced cell death is mediated by
oxidative stress. GSH is an important intracellular antioxidant
that protects cells from damage caused by free radicals, peroxides
and toxins. It is able to remove O2•− and
provide electrons for glutathione peroxidase to reduce
H2O2 to H2O. Apoptotic effects are
inversely comparative to GSH content (33–35).
In the current study, PX-12 increased the percentages of
GSH-depleted cells at 72 h. NAC markedly prevented the depletion of
GSH in PX-12-treated HeLa cells. Furthermore, BSO, which augmented
apoptotic cell death and the loss of MMP in PX-12-treated HeLa
cells, increased GSH depletion in these cells. These results
support the hypothesis that the intracellular GSH content has a
decisive effect on cell death (26,28,34).
However, in the present study, caspase inhibitors marginally
prevented GSH depletion in PX-12-treated HeLa cells. Therefore, the
loss of GSH content appeared to be necessary, but not sufficient to
fully induce apoptosis in PX-12-treated HeLa cells.
In conclusion, to the best of our knowledge, this is
the first study to demonstrate that PX-12 inhibits the growth of
HeLa cells via G2/M phase arrest, as well as apoptosis. This
toxicological effect was associated with intracellular increases in
ROS levels and GSH depletion. The present study provides an
important insight into the toxicological effects of PX-12 on HeLa
cells with respect to ROS and GSH levels.
Acknowledgements
This study was supported by the National Research
Foundation of Korea grant funded by the Korea government (MSIP)
(no. 2008-0062279) and research funds of Chonbuk National
University in 2013.
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
Trx
|
thiore doxin
|
|
GSH
|
glutathione
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-AlaAsp-fluoromethylketone
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-
Asp-Glu-Val-Asp-fluoromethylketone
|
|
Z-IETD-FMK
|
benzyl
oxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
MMP
|
mitochondrial membrane potential
|
|
MTT
|
3-(4,5-dimethylthiazol2-yl)-2,5-diphenyltetrazolium bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
References
|
1
|
Shi Y, Tang B, Yu PW, Tang B, Hao YX, Lei
X, Luo HX and Zeng DZ: Autophagy protects against
oxaliplatin-induced cell death via ER stress and ROS in Caco-2
cells. PLoS One. 7:e510762012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Nino A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lo YL, Wang W and Ho CT:
7,3′,4′-Trihydroxyisoflavone modulates multidrug resistance
transporters and induces apoptosis via production of reactive
oxygen species. Toxicology. 302:221–232. 2012.
|
|
6
|
Bell EL, Emerling BM, Ricoult SJ and
Guarente L: SirT3 suppresses hypoxia inducible factor 1α and tumor
growth by inhibiting mitochondrial ROS production. Oncogene.
30:2986–2996. 2011.PubMed/NCBI
|
|
7
|
Li C, Thompson MA, Tamayo AT, Zuo Z, Lee
J, Vega F, Ford RJ and Pham LV: Over-expression of Thioredoxin-1
mediates growth, survival, and chemoresistance and is a druggable
target in diffuse large B-cell lymphoma. Oncotarget. 3:314–326.
2012.PubMed/NCBI
|
|
8
|
Lim JY, Yoon SO, Hong SW, Kim JW, Choi SH
and Cho JY: Thioredoxin and thioredoxin-interacting protein as
prognostic markers for gastric cancer recurrence. World J
Gastroenterol. 18:5581–5588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang J, Li C, Ding L, Guo Q, You Q and Jin
S: Gambogic acid deactivates cytosolic and mitochondrial
thioredoxins by covalent binding to the functional domain. J Nat
Prod. 75:1108–1116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chae JS, Gil Hwang S, Lim DS and Choi EJ:
Thioredoxin-1 functions as a molecular switch regulating the
oxidative stress-induced activation of MST1. Free Radic Biol Med.
53:2335–2343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ungerstedt J, Du Y, Zhang H, Nair D and
Holmgren A: In vivo redox state of human thioredoxin and redox
shift by the histone deacetylase inhibitor suberoylanilide
hydroxamic acid (SAHA). Free Radic Biol Med. 53:2002–2007. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Lu H, Wang D, Li S, Sun K, Wan X,
Taylor EW and Zhang J: Inhibition of glutathione synthesis
eliminates the adaptive response of ascitic hepatoma 22 cells to
nedaplatin that targets thioredoxin reductase. Toxicol Appl
Pharmacol. 265:342–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Samuel SM, Thirunavukkarasu M, Penumathsa
SV, Koneru S, Zhan L, Maulik G, Sudhakaran PR and Maulik N:
Thioredoxin-1 gene therapy enhances angiogenic signaling and
reduces ventricular remodeling in infarcted myocardium of diabetic
rats. Circulation. 121:1244–1255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fath MA, Ahmad IM, Smith CJ, Spence J and
Spitz DR: Enhancement of carboplatin-mediated lung cancer cell
killing by simultaneous disruption of glutathione and thioredoxin
metabolism. Clin Cancer Res. 17:6206–6217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wondrak GT: Redox-directed cancer
therapeutics: molecular mechanisms and opportunities. Antioxid
Redox Signal. 11:3013–3069. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Welsh SJ, Williams RR, Birmingham A,
Newman DJ, Kirkpatrick DL and Powis G: The thioredoxin redox
inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin
inhibit hypoxia-induced factor 1alpha and vascular endothelial
growth factor formation. Mol Cancer Ther. 2:235–243. 2003.
|
|
17
|
Mukherjee A and Martin SG: The thioredoxin
system: a key target in tumour and endothelial cells. Br J Radiol.
81(Spec No 1): S57–S68. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baker AF, Adab KN, Raghunand N, Chow HH,
Stratton SP, Squire SW, Boice M, Pestano LA, Kirkpatrick DL and
Dragovich T: A phase IB trial of 24-hour intravenous PX-12, a
thioredoxin-1 inhibitor, in patients with advanced gastrointestinal
cancers. Invest New Drugs. 31:631–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ramanathan RK, Kirkpatrick DL, Belani CP,
Friedland D, Green SB, Chow HH, Cordova CA, Stratton SP, Sharlow
ER, Baker A and Dragovich T: A Phase I pharmacokinetic and
pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1,
in patients with advanced solid tumors. Clin Cancer Res.
13:2109–2114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hedley D, Pintilie M, Woo J, Nicklee T,
Morrison A, Birle D, Fyles A, Milosevic M and Hill R: Up-regulation
of the redox mediators thioredoxin and apurinic/apyrimidinic
excision (APE)/Ref-1 in hypoxic microregions of invasive cervical
carcinomas, mapped using multispectral, wide-field fluorescence
image analysis. Am J Pathol. 164:557–565. 2004. View Article : Google Scholar
|
|
21
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han YH and Park WH: The effects of
N-acetyl cysteine, buthionine sulfoximine, diethyldithiocarbamate
or 3-amino-1,2,4-triazole on antimycin A-treated Calu-6 lung cells
in relation to cell growth, reactive oxygen species and
glutathione. Oncol Rep. 22:385–391. 2009.
|
|
23
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of carbonyl cyanide p-(trifluoromethoxy)
phenylhydrazone on the growth inhibition in human pulmonary
adenocarcinoma Calu-6 cells. Toxicology. 265:101–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of human lung cancer Calu-6 cells
via arresting the cell cycle arrest. Toxicol In Vitro.
22:1605–1609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
26
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*−) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Griffiths EJ: Mitochondria - potential
role in cell life and death. Cardiovasc Res. 46:24–27. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vogt A, Tamura K, Watson S and Lazo JS:
Antitumor imidazolyl disulfide IV-2 causes irreversible G(2)/M cell
cycle arrest without hyperphosphorylation of cyclin-dependent
kinase Cdk1. J Pharmacol Exp Ther. 294:1070–1075. 2000.PubMed/NCBI
|
|
31
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee YJ, Kim JH, Chen J and Song JJ:
Enhancement of metabolic oxidative stress-induced cytotoxicity by
the thioredoxin inhibitor 1-methylpropyl 2-imidazolyl disulfide is
mediated through the ASK1-SEK1-JNK1 pathway. Mol Pharmacol.
62:1409–1417. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
34
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
35
|
Han YH, Kim SZ, Kim SH and Park WH:
Suppression of arsenic trioxide-induced apoptosis in HeLa cells by
N-acetylcysteine. Mol Cells. 26:18–25. 2008.PubMed/NCBI
|

















