Introduction
Cancer development depends on a complex tissue
environment for growth and metastasis (1). It is now widely recognized that immune
reaction and vascular growth support tumor growth (2). Since that breakthrough,
anti-angiogenic therapy has been successfully introduced in
clinical cancer therapy to starve tumors (3–6).
Theoretically, using anti-angiogenic agents to block
factors crucial to tumor angiogenesis should offer several
advantages over conventional chemotherapy agents. First,
anti-angiogenic therapy can treat all solid tumors without being
restricted to a specific tumor type. Second, as anti-angiogenic
therapy targets the endothelial cells within the tumor vasculature,
the agents within the blood stream directly affect the targeted
cells, without penetration of the tumor being necessary.
Furthermore, anti-angiogenic drugs are not expected to give rise to
drug resistance as they do not target the highly mutable cancer
cell population, but rather the more genetically stable endothelial
cells (7,8). Anti-angiogenic therapy should thus
allow for prolonged treatment with anti-angiogenic drugs, without
giving rise to resistance.
Currently, hundreds of clinical trials involving
anti-angiogenic agents are underway. Despite the initial promising
performance of anti-angiogenic drugs in clinical trials,
anti-angiogenesis therapy faces numerous challenges, including
inherent and acquired resistance. The majority of cancer patients
eventually demonstrate a lack of response to anti-angiogenic
therapy while on the treatment regimen (4–6).
Studies using clinical and preclinical models have documented the
involvement of certain molecular and cellular mechanisms (9–11). At
present, the proposed mechanisms include alternative angiogenic
pathways, such as selective pressure of hypoxia, cancer stem cells,
autophagy, recruitment of vascular progenitors and modulators, and
tumor dormancy (11). In
particular, previous studies indicate that acquired drug resistance
in tumor endothelial cells is involved in drug resistance in cancer
patients (12,13).
Multidrug resistance is considered a major obstacle
for successful chemotherapy in the treatment of cancer.
Chemotherapy loses effectiveness over time. One reason is the drug
resistance caused by the compensatory response of tumor cells
(10,14). One of the main underlying mechanisms
for multidrug resistance in cancer chemotherapy is the
overproduction of ATP-binding cassette (ABC) transporter, which
serves as a pump to remove toxic drugs from tumor cells, thus
rendering the tumor cells resistant to multiple chemotherapeutic
drugs.
Clinical studies of human cancer have found a
correlation between P-glycoprotein (P-gp) overexpression in tumor
tissues with decreased survival and poor prognosis (14). High P-gp expression has been found
in tumor endothelial cells, likely in response to vascular
endothelial growth factor stimulation (15). We have also shown that the
chemotherapeutic agent doxorubicin (Dox) induces high levels of
P-gp in endothelial cells (12).
Our previous study established two endothelial cell lines, HMECd1
and HMECd2, that exhibited high drug resistance to doxorubicin
(Dox) induction in vitro. These two stabilized sub cell
lines demonstrated 15- and 24-fold increases in resistance to Dox.
Acquired drug resistance in endothelial cells was also revealed to
attenuate the efficacy of doxorubicin treatment in a mouse tumor
model. Dox-induced drug resistance in these endothelial cells was
predominantly due to MDR1/P-gp upregulation. Inhibiting the
activity of P-gp could reverse the resistance of endothelial cells.
Furthermore, the drug resistance of endothelial cells attenuated
the efficacy of doxorubicin treatment in vivo (12). This previous study indicated that
the acquired drug resistance of tumor vessels plays a critical role
in cancer therapy.
The breast cancer resistance protein (ABCG2) is
another ABC transporter that has been identified as a molecular
cause of multidrug resistance in diverse cancer cells (16,17).
As an efflux transporter for xenobiotics and unwanted toxic
compounds, ABCG2 has been characterized as an important component
of self-defense systems in organisms (18). In the brain microvasculature, ABCG2
is located on the luminal surface of microvessel endothelium and
hence may constitute an important component of the blood-brain
barrier (19).
Sunitinib is an oral multi-targeted receptor
tyrosine kinase inhibitor of vascular endothelial growth-factor
receptors (20,21). Currently, sunitinib is used to treat
advanced or metastatic renal cell carcinoma, gastrointestinal
stromal tumors, meningioma and pancreatic neuroendocrine tumors.
Clinical trials of combined sunitinib therapy with chemotherapy are
ongoing (22–24). Patient resistance to sunitinib
treatment has been reported (11,25,26).
The aim of the present study was to investigate the risk of
acquired and cross-resistance to anti-angiogenic drugs in
endothelial cells during chemotherapy.
Materials and methods
Materials
Mouse monoclonal anti-P-gp, anti-ABCG2 and anti-MRP1
antibodies were purchased from Abcam (Cambridge, UK). Sunitinib was
obtained from Pfizer, Inc. (New York, NY, USA). Doxorubicin
chlorhydrate was purchased from Amersham Pharmacia Biotech, Inc.
(Uppsala, Sweden). Verapamil was obtained from Calbiochem
(Billerica, MA, USA). Paclitaxel, vinblastine, cyclosporine A,
fumitremorgin C, diethylstilbestrol and MK571 were purchased from
Sigma-Aldrich (Saint Louis, MO, USA).
Cell culture
Parental and resistant HMEC-1 cell lines, obtained
from Dr TL Lawley (Department of Dermatology, Atlanta, GA, USA),
were cultured in MCDB-131 medium supplemented with 10% fetal calf
serum (FCS), 2 mM L-glutamine, 10 ng/ml epidermal growth factor, 1
μg/ml hydrocortisone, 100 units/ml penicillin, and 100 μg/ml
streptomycin, as described elsewhere (12,27).
Dox-resistant HMEC cells were obtained by continuously exposing
cells to increasing concentrations of Dox, between 0.001 and 0.24
μg/ml, over a 12-week period, as previously described (12). Two sub cell lines of HMEC-1 cells
were collected, HMECd1 cells were maintained in a culture with 0.08
μg/ml Dox and HMECd2 cells were maintained in 0.24 μg/ml Dox. No
mutagenic agents were used to establish these Dox-resistant HMEC
cells. To observe the reversibility of the drug resistance of the
cells, Dox was withdrawn from the culture medium of HMECd1 and
HMECd2 cells. All cell types were digested with trypsin-EDTA once
or twice a week and cultured in a 37°C incubator with a 100%
humidified atmosphere of 5% CO2.
MTS cell proliferation assay
Cell viability was determined using MTS cell
proliferation assay (Promega, Madison, WI, USA). Cells grew to a
confluence of 90% in 75 cm2 cell culture flasks and were
passed into 96-well plates (7,500 cells/well). Each well contained
100 μl of culture medium, which was supplemented with various
concentrations of drugs or with a concentration of dimethyl
sulfoxide as a control. Following incubation for either 24, 48 or
72 h, 20 μl of the MTS reagent was added to each well and the plate
was placed in the 5% CO2 incubator at 37°C for an
additional 2 h. The optical density (OD) was then read at 492 nm
using a microplate reader (Labsystems Multiskan MS; MTX Lab Systems
Inc., Vienna, VA, USA). The half maximal inhibitory concentration
(IC50) values were defined as the concentration of drug
producing 50% inhibition of cell growth and the resistance index
corresponding to the ratio of IC50 values between the
resistant and parental cell lines. Experiments were performed in
triplicate and repeated at least three times.
Blocking effect assay
The experiments used ABCG2 inhibitors, 5 μM
fumitremorgin C and 0.5 μM diethylstilbestrol, and P-gp inhibitors,
2.5 μM cyclosporine A, 1 μM verapamil and 5 μM MK571. Following
incubation for 48 or 72 h, the cell viability was assessed using an
MTS assay. The reversal fold (RF) values, a measure of the potency
of reversal, were obtained by fitting the data to RF =
IC50 of cytotoxic drug alone/IC50 of
cytotoxic drug in the presence of a modulator (28).
Evaluation of mRNA expression via
quantitative polymerase chain reaction (qPCR)
The HMEC-1, HMECd1 and HMECd2 cells were treated
with 2.5 μM cyclosporine A, 1 μM verapamil, 5 μM fumitremorgin C,
0.5 μM diethylstilbestrol or 5 μM MK571 for 24 h. Subsequent to
incubation, the treated and non-treated cells were harvested, and
total RNA was prepared using the SV total RNA isolation system kit
(Promega). The purity of total RNA was checked by a ratio of
A260/A280 (>1.9). Total RNA (50 ng) was
used to synthesize the first-strand cDNA in a 20-μl reaction
solution using the GoScript Reverse Transcription System kit
(Promega). Then, 2 μl of cDNA was used for qPCR in triplicates
using a TaqMan® gene expression assay (Applied
Biosystems, Foster City, CA, USA) and the primers for P-gp
(Hs01067802_m1), ABCG2 (Hs01053790_m1), multidrug resistance
protein (MRP) 1 (Hs00219905_m1), as well as the primers for TATA
box binding protein (TBP) as controls (Hs99999910_m1; Applied
Biosystems). qPCR was performed by 10 min of initial denaturation
followed by 44 cycles of 15 sec at 95°C and 60 sec at 60°C in a
BioRad CFX96® Real-time system (Bio-Rad Laboratories, Hercules, CA,
USA). The ΔCt method was used to analyze the qPCR results, and TBP
was used as an internal control for mRNA-level normalization.
Evaluation of protein expression using
western blot analysis
Western blot analysis was performed on whole-cell
lysates by incubating the cells in the lysis buffer (10 mM Tris pH
6.8, 1 mM EDTA, 10% Nonidet P-40, 1 mM phenylmethanesulfonyl
fluoride, 0.1% SDS) on ice for 30 min. Cell debris was removed by
centrifugation at 16,000 × g for 10 min. Protein concentration was
determined by bicinchoninic acid protein assay (Thermo Fisher
Scientific, Waltham, MA, USA). A 50 μg protein sample from each
sample was loaded on an 8% SDS-PAGE gel, and the protein was
transferred to a polyvinylidene fluoride membrane using the iBlot
dry blotting system (Invitrogen, Carlsbad, CA, USA). The membranes
were blocked with 5% non-fat dry milk for 1 h and incubated with
either anti-P-gp (ab-3364; 1:20; Abcam), anti-MRP1 (ab-32574;
1:250; Abcam) or anti-ABCG2 antibodies (ab-3380; 1:100; Abcam) at
4°C overnight. The membranes were then washed with a Tris buffered
saline with Tween 20 buffer for 1 h and incubated with the
appropriate horseradish peroxidase-conjugated secondary antibodies
(Invitrogen), diluted in blocking buffer, for 1 h at room
temperature. Subsequent to washing, western blotting luminol
reagent (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was added
to the membranes and the chemiluminescence was recorded using a
Fuji LAS-3000 system (Fujifilm, Tokyo, Japan). The membranes were
then treated with an antibody-stripping buffer (Gene
Bio-Application Ltd., Kfar Hanagid, Israel) and incubated with
anti-actin antibody (1:4,000 dilution; Sigma-Aldrich) as a
control.
Statistical analyses
The data were analyzed using one-way analysis of
variance and Mann-Whitney U tests, as appropriate. The qPCR data is
presented as the mean ± standard error of the mean. The remaining
data is presented as the mean ± standard deviation. P≤0.05 was
considered to indicate a statistically significant difference.
Results
Endothelial cells resistant to
anti-angiogenesis drugs
To address the question of the response of HMECd1
and HMECd2 to the anti-angiogenic drugs, the efficacy of sunitinib
was tested in vitro. The first experiment with MTS assay
revealed that HMEC-1 cells are initially sensitive to sunitinib
treatment. However, compared with their parental cells, HMECd1 and
HMECd2 cells revealed 2.1- and 4.51-fold increases in
drug-resistance to sunitinib (Table
I). The increase in sunitinib resistance accompanied the
increase in Dox resistance (Table
I). This observation corresponded with the typical multi-drug
resistance of endothelial cells in response to Dox induction.
 | Table IDox-induced cross-resistance to Dox
and sunitinib in HMEC-1 endothelial cells. |
Table I
Dox-induced cross-resistance to Dox
and sunitinib in HMEC-1 endothelial cells.
| HMEC-1 | HMECd1 | HMECd2 |
|---|
|
|
|
|
|---|
| Agents | IC50,
μM | IC50,
μM | RI | IC50,
μM | RI |
|---|
| Sunitinib | 4.271±0.501 | 8.585±0.642 | 2.01a | 19.252±0.855 | 4.51a |
| Doxorubicin | 0.056±0.006 | 0.812±0.050 | 14.50a | 1.209±0.085 | 21.60a |
ABCG2 and P-gp are predominantly
expressed in the resistant endothelial cells
qPCR was used to measure changes in drug efflux
transporter gene expression in the Dox-induced resistant
endothelial cells. The P-gp and ABCG2 expression in HMECd1 and
HMECd2 cells increased significantly compared to parental cells
(1.41- and 1.68-fold for ABCG2; 3.4- and 7.2-fold for P-gp;
Fig. 1). To evaluate the influence
of the ABC transporter blockers used in the study of gene
expression, the changes in P-gp, ABCG2 and MRP1 mRNA levels in the
presence of the inhibitors of the three transporters were
quantified, respectively.
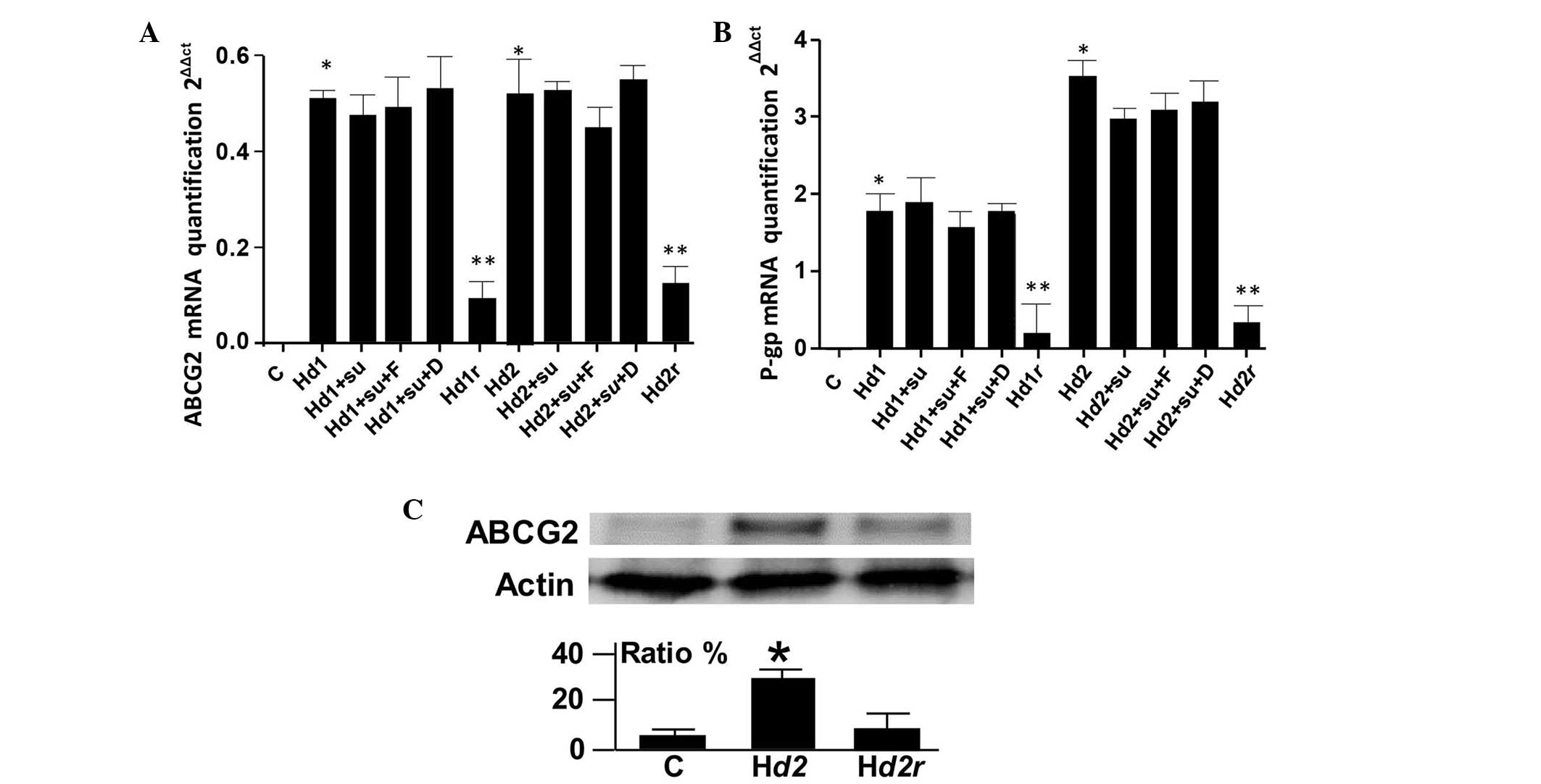 | Figure 1Induced ABCG2 expression in HMEC-1
endothelial cells. (A) qPCR (primer, Hs01053790_m1) of ABCG2 mRNA
levels in treated or non-treated HMEC-1, HMECd1, HMECd2, HMECd1r
and HMECd2r cells cultured without Dox for three weeks. Sunitinib,
fumitremorgin C and diethylstilbestrol were used to treat the
cells. The results were obtained from three independent
experiments. *P<0.05, vs. non-treated cells. (B) qPCR
(primer, Hs01067802_m1) of P-gp mRNA levels in treated or
non-treated HMEC-1, HMECd1, HMECd2 and HMECd2r cells. Sunitinib,
fumitremorgin C, and diethylstilbestrol were used to treat the
cells. The results were obtained from three independent
experiments. *P<0.05 and **P<0.01, vs.
non-treated cells. (C) Western blot analysis of ABCG2 levels in
HMEC-1, HMECd2 and HMECd2r cells. The data for the ratio were
obtained from three repeated blots. *P<0.05, vs. the
control and Hd2r cells. C, HMEC-1; Hd1, HMECd1; Hd2, HMECd2; Hd1r,
HMECd1r; Hd2r, HMECd2r; Su, sunitinib; F, fumitremorgin C; D,
diethylstilbestrol; ABCG2, breast cancer resistance protein. |
There was no significant change in the gene
expression of P-gp and ABCG2 in HMECd1 and HMECd2 cells when the
function of P-gp or ABCG2 was blocked (Fig. 1A and B). The qPCR results also
indicated that MRP1 was not induced in HMECd1 and HMECd2 cells
(data not shown). Western blotting revealed approximately two- to
four-fold increases in the ABCG2 protein expression of the cells.
Withdrawal of Dox in the culture media for more than three weeks
resulted in a decrease in P-gp and ABCG2 expression in HMECd1
(Hd1r)and HMECd2 (Hd2r) cells (Fig.
1). This indicated that the P-gp and ABCG2 expression was
reversible.
Blocking ABCG2 attenuates the resistance
of Dox-induced endothelial cells to sunitinib
The survival of HMECd1 and HMECd2 cells was examined
subsequent to sunitinib treatment in the presence of the P-gp
blockers cyclosporine A and verapamil, or in the presence of the
ABCG2 blockers fumitremorgin C and diethylstilbestrol. The results
revealed that only the blockade of ABCG2 function significantly
restored the sensitivity of Dox-induced of endothelial cells to
sunitinib (Table II). By contrast,
the P-gp inhibitors demonstrated no such effect.
| Table IIEffect of P-gp inhibitors and ABCG2
inhibitors on sunitinib resistance in HMEC-1 cells. |
Table II
Effect of P-gp inhibitors and ABCG2
inhibitors on sunitinib resistance in HMEC-1 cells.
| HMEC-1 | HMECd1 | HMECd2 |
|---|
|
|
|
|
|---|
| Agents | IC50,
μM | IC50,
μM | RI | RF | IC50,
μM | RI | RF |
|---|
| Sunitinib | 4.271±0.501 | 8.585±0.642 | 2.01a | 1.00 | 19.252±0.855 | 4.51a | 1.00 |
| + 5 μM fumC | 4.359±0.622 | 5.886±0.417 | 1.35b | 1.45 | 6.163±0.062 | 1.41b | 3.12 |
| + 0.5 μM die | 4.204±0.468 | 6.259±0.541 | 1.48b | 1.37 | 7.159±0.057 | 1.70b | 2.69 |
| + 1 μM vrp | 4.952±0.875 | 9.159±0.356 | 1.84 | 0.94 | 17.659±0.526 | 3.57 | 1.09 |
| + 2.5 μM cysA | 4.098±0.562 | 8.871±0.459 | 2.16 | 0.97 | 20.348±0.328 | 4.97 | 0.95 |
Discussion
Drug resistance remains a difficult, unsolved issue
in cancer therapy. Since the start of the use of anti-angiogenic
therapy, it was expected to be an exception to drug resistance
(7,8). However, primary or acquired drug
resistance was soon reported in anti-angiogenic therapy and the
mechanisms of that resistance have been periodically reviewed
(9–11). In a previous study, it was
demonstrated that doxorubicin successfully induced multiple
resistance in endothelial cells (12). In the present study, the response of
two stabilized endothelial cell lines, HMECd1 and HMECd2, to the
anti-angiogenesis drug sunitinib was evaluated.
The present study provides evidence that ABCG2
mediates the substrate efflux of sunitinib in endothelial cells.
The present study demonstrated that, in addition to P-gp
upregulation, ABCG2 expression was upregulated in HMECd1 and HMEd2
cells. The ABCG2 protein levels, revealed by western blot analysis,
were found to possess a two- to four-fold increase in HMECd1 and
HMECd2 cells compared with their parental cells. Similarly, qPCR
revealed 1.41- and 1.68-fold increases in ABCG2 gene expression in
the HMECd1 and HMECd2 cells. It was revealed that P-gp was not
involved in sunitinib resistance as two inhibitors of P-gp,
cyclosporine A and verapamil, failed to reverse sunitinib
resistance in HMECd1 and HMEC2. By contrast, the blockade of ABCG2
activity by fumitremorgin C and diethylstilbestrol greatly
inhibited the capacity of the cells for sunitinib resistance. These
results indicate that ABCG2 plays a major functional role in the
resistance of HMECd1 and HMEC2 endothelial cells to sunitinib in
vitro. The present results were in agreement with previous
reports that revealed the involvement of ABCG2 in the resistance of
cells to sunitinib treatment, although the involvement of P-gp has
not been excluded in in vivo studies (29–31).
As resistance to sunitinib in endothelial cells can be induced by
Dox, potential cross-resistance in combined therapy that uses
chemotherapy and targeted therapy may occur in clinical trials.
Cross-resistance in cancer therapy was observed in
clinical settings >50 years ago (32,33).
Currently, as the combined use of chemotherapy and anti-angiogenic
drugs develops rapidly, it is particularly important to explore
cross-resistance. Indeed, clinical trials evaluating the combined
use of targeted therapy and chemotherapy are extremely dynamic. For
example, >10 chemotherapy drugs, including cisplatin,
5-fluoroutacil and paclitaxel are currently being explored in
combination with sunitinib in clinical studies (22–24).
Furthermore, in the present study, resistance of the endothelial
cells to other agents used in targeted therapy was observed (data
not shown). Ongoing clinical trials comprise numerous
anti-angiogenic drugs (34,35). These trials require additional
knowledge about the occurrence of cross-resistance, not only in
tumor cells but also in endothelial cells.
Cross-resistance is complex as each ABC transporter
can induce the efflux of a panel of chemical molecules based on
physical affinity (18).
Furthermore, a drug that consists of a single chemical can induce
the upregulation of more than one ABC protein, as described in
previous studies (12,20). During the establishment and
optimization of a clinical therapeutic protocol with the provided
therapeutic targets, it would be beneficial to evaluate and
consider the potential risk of the development of cross-resistance
to the treatment drugs. From this standpoint, additional effort
would aid in the optimization of the combined use of
chemotherapeutic agents and anti-angiogenic agents. Notably, as
cross-resistance may occur in tumor and endothelial cells, it can
also be speculated that the biological properties of drug
resistance in these two types of cells may exhibit differences.
Since anti-angiogenic agents have been used
clinically, patient resistance to anti-angiogenic drugs has been
reported and analyzed frequently. Proposed mechanisms of resistance
include alternative angiogenic escape factors, an increase in the
stem cell population that is resistant to hypoxia, the selection of
cells with acquired metastatic and invasive potential by hypoxia
and tumor cell dormancy (11).
Avoiding cross-resistance is expected to contribute to the further
improvement of anticancer therapy, together with strategies that
target multiple pathways involved in angiogenesis and
resistance.
Acknowledgements
The authors thank the Institute of Cancer (INCA,
PL06_130) and the Association Ti’toine for their support and also
thank Professors Soria J and Soria C for their support.
Abbreviations:
|
P-gp
|
P-glycoprotein
|
|
MRP1
|
multidrug resistance associated
proteins
|
|
ABCG2
|
breast cancer resistance protein
|
|
qPCR
|
quantitative polymerase chain
reaction
|
References
|
1
|
Folkman J: Tumor angiogenesis: therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Husein B, Abdalla M, Trepte M, Deremer
DL and Somanath PR: Antiangiogenic therapy for cancer: an update.
Pharmacotherapy. 32:1095–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Young RJ and Reed MW: Anti-angiogenic
therapy: concept to clinic. Microcirculation. 19:115–125. 2012.
View Article : Google Scholar
|
|
6
|
Wu JM and Staton CA: Anti-angiogenic drug
discovery: lessons from the past and thoughts for the future.
Expert Opin Drug Discov. 7:723–743. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kerbel RS: Inhibition of tumor
angiogenesis as a strategy to circumvent acquired resistance to
anti-cancer therapeutic agents. Bioessays. 13:31–36. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boehm T, Folkman J, Browder T and O’Reilly
MS: Antiangiogenic therapy of experimental cancer does not induce
acquired drug resistance. Nature. 390:404–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sweeney CJ, Miller KD and Sledge GW Jr:
Resistance in the anti-angiogenic era: nay-saying or a word of
caution? Trends Mol Med. 9:24–29. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Broxterman HJ, Lankelma J and Hoekman K:
Resistance to cytotoxic and anti-angiogenic anticancer agents:
similarities and differences. Drug Resist Updat. 6:111–127. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giuliano S and Pagès G: Mechanisms of
resistance to anti-angiogenesis therapies. Biochimie. 95:1110–1119.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang L, Perrault C, Coelho-Martins J, et
al: Induction of acquired drug resistance in endothelial cells and
its involvement in anticancer therapy. J Hematol Oncol. 6:492013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hida K, Akiyama K, Ohga N, Maishi N and
Hida Y: Tumour endothelial cells acquire drug resistance in a
tumour microenvironment. J Biochem. 153:243–249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pakos EE and Ioannidis JP: The association
of P-glycoprotein with response to chemotherapy and clinical
outcome in patients with osteosarcoma. A meta-analysis. Cancer.
98:581–589. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akiyama K, Ohga N, Hida Y, et al: Tumor
endothelial cells acquire drug resistance by MDR1 up-regulation via
VEGF signaling in tumor microenvironment. Am J Pathol.
180:1283–1293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lage H and Dietel M: Effect of the
breast-cancer resistance protein on atypical multidrug resistance.
Lancet Oncol. 1:169–175. 2000. View Article : Google Scholar
|
|
17
|
Xu J, Peng H and Zhang JT: Human multidrug
transporter ABCG2, a target for sensitizing drug resistance in
cancer chemotherapy. Curr Med Chem. 14:689–701. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ni Z, Bikadi Z, Rosenberg MF and Mao Q:
Structure and function of the human breast cancer resistance
protein (BCRP/ABCG2). Curr Drug Metab. 11:603–617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cooray HC, Blackmore CG, Maskell L and
Barrand MA: Localisation of breast cancer resistance protein in
microvessel endothelium of human brain. Neuroreport. 13:2059–2063.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mena AC, Pulido EG and Guillén-Ponce C:
Understanding the molecular-based mechanism of action of the
tyrosine kinase inhibitor: sunitinib. Anticancer Drugs. 21(Suppl
1): S3–S11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shojaei F: Anti-angiogenesis therapy in
cancer: current challenges and future perspectives. Cancer Lett.
320:130–137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmitt JM, Sommers SR, Fisher W, et al:
Sunitinib plus paclitaxel in patients with advanced esophageal
cancer: a phase II study from the Hoosier Oncology Group. J Thorac
Oncol. 7:760–763. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galsky MD, Hahn NM, Powles T, et al:
Gemcitabine, Cisplatin, and sunitinib for metastatic urothelial
carcinoma and as preoperative therapy for muscle-invasive bladder
cancer. Clin Genitourin Cancer. 11:175–181. 2013. View Article : Google Scholar
|
|
24
|
Gómez-Martín C, Salazar R, Montagut C, et
al: A phase I, dose-finding study of sunitinib combined with
cisplatin and 5-fluorouracil in patients with advanced gastric
cancer. Invest New Drugs. 31:390–398. 2013. View Article : Google Scholar
|
|
25
|
Wang WL, Conley A, Reynoso D, Nolden L,
Lazar AJ, George S and Trent JC: Mechanisms of resistance to
imatinib and sunitinib in gastrointestinal stromal tumor. Cancer
Chemother Pharmacol. 67(Suppl 1): S15–S24. 2011. View Article : Google Scholar
|
|
26
|
Bracci R, Maccaroni E and Cascinu S:
Transient sunitinib resistance in gastrointestinal stromal tumors.
N Engl J Med. 368:2042–2043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Trochon-Joseph V, Martel-Renoir D, Mir LM,
et al: Evidence of antiangiogenic and antimetastatic activities of
the recombinant disintegrin domain of metargidin. Cancer Res.
64:2062–2069. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji BS, He L and Liu GQ: Reversal of
p-glycoprotein-mediated multidrug resistance by CJX1, an amlodipine
derivative, in doxorubicin-resistant human myelogenous leukemia
(K562/DOX) cells. Life Sci. w77:2221–2232. 2005. View Article : Google Scholar
|
|
29
|
Kawahara H, Noguchi K, Katayama K,
Mitsuhashi J and Sugimoto Y: Pharmacological interaction with
sunitinib is abolished by a germ-line mutation (1291T>C) of
BCRP/ABCG2 gene. Cancer Sci. 101:1493–1500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizuno T, Fukudo M, Terada T, et al:
Impact of genetic variation in breast cancer resistance protein
(BCRP/ABCG2) on sunitinib pharmacokinetics. Drug Metab
Pharmacokinet. 27:631–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kunimatsu S, Mizuno T, Fukudo M and
Katsura T: Effect of P-glycoprotein and breast cancer resistance
protein inhibition on the pharmacokinetics of sunitinib in rats.
Drug Metab Dispos. 41:1592–1597. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hutchison DJ: Cross resistance and
collateral sensitivity studies in cancer chemotherapy. Adv Cancer
Res. 7:235–250. 1963. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hutchison DJ: Conference on obstacles to
the control of acute leukemia. Studies on cross-resistance and
collateral sensitivity (1962–1964). Cancer Res. 25:1581–1595.
1965.PubMed/NCBI
|
|
34
|
Cesca M, Bizzaro F, Zucchetti M and
Giavazzi R: Tumor delivery of chemotherapy combined with inhibitors
of angiogenesis and vascular targeting agents. Front Oncol.
3:2592013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu XY, Ma W, Gurung K and Guo CH:
Mechanisms of tumor resistance to small-molecule vascular
disrupting agents: treatment and rationale of combination therapy.
J Formos Med Assoc. 112:115–124. 2013. View Article : Google Scholar : PubMed/NCBI
|














