Introduction
Tumors of the pineal region (PR) are rare. The
tumors are most commonly found in children and infants, while only
1% occur during adulthood (1,2). The appropriate pathological
classification and grading of the malignancy of tumors of the PR is
essential for determining the clinical management and prognosis
(3,4).
Tumors of the PR can be subdivided into four main
histomorphological groups: Pineal-parenchymal tumors (PPT), germ
cell tumors (GCT), glial tumors and miscellaneous tumors.
PPTs originating from the pineal gland itself
account for 14–27% of PR tumors (1,5–7). In addition to grade I pineocytoma and
grade IV pineoblastoma, PPT of intermediate differentiation (PPTID)
was recognized in the 2007 World Health Organization (WHO)
classification as an intermediate-grade malignancy (II or III).
Therefore, these tumors may have been summarized in the past under
grade I pineocytoma or grade IV pineoblastoma (5). The origin of pineal cysts remains
unclear, but they are believed to be non-neoplastic (8). GCTs are among the most abundant entities
(40%) in the PR, and include germinomas and non-germinomatous
tumors, such as epidermoid cysts, teratomas and yolk sac tumors
(5,9,10). Glial
tumors, most commonly grade I pilocytic astrocytomas, account for
up to 25% of tumors of the PR (5,11).
Miscellaneous tumors of the PR include solitary fibrous tumors
(SFT) (12) and metastases. Rarely,
other brain tumors, including meningioma, glioblastoma, ependymoma,
plexus papilloma and neuroendocrine tumors (NETs), can also occur
in the PR (13–16).
The early diagnosis of tumors in the PR is often
delayed through unspecific clinical symptoms. Imaging studies can
be suggestive of the type of tumor of the PR, but only occasionally
provide the exact diagnosis (9,17,18). Serum and cerebrospinal fluid markers
have been the most useful in the pre-operative evaluation, as
α-fetoprotein (AFP) and β-human chorionic gonadotropin (β-HCG) are
indicative of malignant GCT (19).
Nevertheless, the absence of AFP or β-HCG does not rule out a
malignant GCT (20). Markers of PPTs
are not as well characterized as their germ cell counterparts, and
include melatonin and the S-antigen, neither of which have proven
valuable in establishing the correct diagnosis (21,22).
Additionally, the histopathological diagnosis of tumors of the PR
is often difficult due to the inherently small size of the biopsies
for diagnosis and the wide range of histological tumor entities in
this brain region (3,5). Immunohistochemical support is not always
decisive to identify the histomorphological diversity of tumors of
the PR (1,11,23).
Therefore, valuable diagnostic markers have to be
defined to allow a histogenetically-based diagnosis. Recently, we
described a chromosomal pattern, which can differentiate papillary
tumors of the PR from other papillary tumors of the PR (24). Specific genetic changes in other
tumors of the PR are yet to be determined. The present study aimed
to characterize tumors of the PR using comparative genomic
hybridization (CGH). In addition to chromosomal aberrations in PPT
and different GCTs of the PR, the present study describes, for the
first time, chromosomal changes in a few rare entities of the
PR.
Materials and methods
Patients
This study included 18 patients in whom tumors of
the PR were primarily surgically resected without any previous
irradiation or chemotherapy between 1997 and 2005. Inclusion
criteria comprised follow-up data and successful cytogenetic
analyses of the tumors. The study was performed with the patients'
informed consent, and according to the guidelines and approval of
the Local Ethics Committee (No. 12/11/10) of the Georg-August
University Göttingen (Göttingen, Germany).
DNA preparation and CGH
The specimens were trimmed to ensure a minimum of
75% tumor cells in the sample. Tumor DNA was extracted from
formalin-fixed and paraffin-embedded tumors of the PR by proteinase
K digestion (2 mg/ml final concentration; Roche Diagnostics GmbH,
Mannheim, Germany) followed by spin column purification (Qiagen
GmbH, Hilden, Germany). The labeling of tumor DNA (nick
translation) was performed with biotin-16-dUTP and normal reference
DNA with digoxigenin-11-dUTP (both Roche Diagnostics GmbH). The
denatured DNA probe containing 2 µg tumor DNA, 1.5 µg reference DNA
and 80 µg COT-1 DNA was hybridized for 3 days to normal metaphase
spreads on glass slides (15×15-mm cover glass area). The slides
were then washed, blocked with bovine serum albumin solution and
incubated with anti-biotin fluorescein-conjugated avidin (Vector
Laboratories Inc., Burlingame, CA, USA) and rhodamine-conjugated
anti-digoxigenin (Roche Diagnostics GmbH) antibodies. Finally, the
slides were washed and mounted in antifade solution (Vector
Laboratories Inc.) containing 2.5 µg/ml
4′,6-diamidino-2-phenylindole (DAPI) counterstain.
Imaging and image analysis
Image acquisition was performed on a Zeiss Axioskop
fluorescence microscope (Zeiss, Göttingen, Germany) equipped with
three separate bandpass filters (a DAPI bandpass, a green single
bandpass and a red single bandpass) and a high sensitivity
monochrome charge coupled device camera (Photometrics, Tuscon, AZ,
USA). For each analysis, the averaged chromosome-specific
green-to-red fluorescence ratios and their 95% confidence intervals
from at least 10 well-selected metaphases were plotted using the
Quips CGH software (Applied Imaging, Newcastle, UK). A gain of DNA
sequences was recorded at chromosomal regions where the
hybridization resulted in a tumor-to-normal ratio of 1.2.
Overrepresentations were considered amplifications when the
fluorescence ratio values were >1.5 in a subregion of a
chromosome arm. A loss of DNA sequences was recorded where the
tumor-to-normal ratio was 0.8. As an internal control, normal
reference DNA was chosen from the opposite gender to ensure correct
technical analyses. The chromosomal regions 1p32-pter, 13p, 14p,
15p, 19, 21p and 22p, and the known constitutive heterochromatic
regions at 1q, 9q, 16q and Yq, and telomeric regions, were excluded
from the analysis.
Statistical analysis
To evaluate chromosomal imbalance distribution in
relation to diagnostic assignment, for each of the entities in the
dataset, gain and loss frequencies were calculated. Copy number
profiles were compared by generating a heatmap of gain and loss
distributions. All statistical analyses are intended to be
exploratory rather than confirmatory due to the relatively low
number of patients per group. P<0.05 was considered to indicate
a statistically significant difference and were determined using
the Student's t-test. Mean values and standard deviations are
shown. Statistical analyses were performed using GraphPadPrism 5.0
software (GraphPad Software Inc., La Jolla, CA, USA).
Results
Clinicopathological data
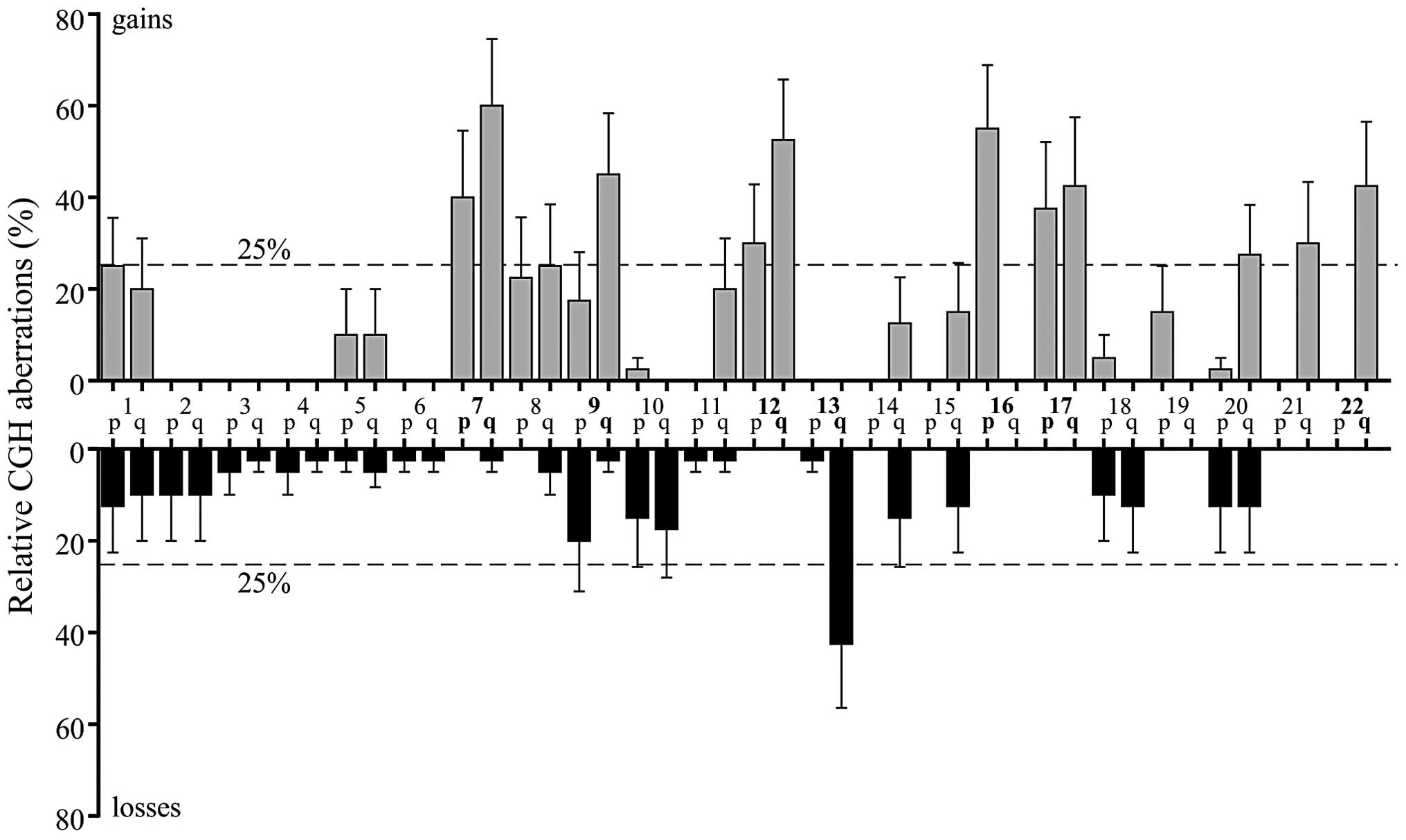
The 18 tumors of the PR included 6 PPTs, 1 pineal
cyst, 6 pineal GCTs and 5 miscellaneous tumors (Table I; Fig.
1). Overall, 83.3% of the patients were male, with a mean age
at the time of tumor diagnosis of 26.6±17.0 years. The male
patients were significantly younger at the time of diagnosis
(22.8±3.7 years; range, 5 months-47 years) than the female patients
(45.4±10.9 years; range, 24–60 years; P<0.035).
 | Table I.Chromosomal aberrations detected in 18
tumors of the pineal region. |
Table I.
Chromosomal aberrations detected in 18
tumors of the pineal region.
|
|
|
|
| CGH aberrations |
|---|
|
|
|
|
|
|
|---|
| Case | Diagnosis | Gender | Age at diagnosis | Gains | Losses |
|---|
| 1 | Pineal cyst | Male | 36 years | +16p |
|
| 2 | PPTID WHO grade
II | Male | 22 years | +1p, +7p,
+7q, +9p, +9q, +11q, +12p, +12q, +15q,
+16p, +18p, +20q, +21q | −10p, −10q,
-13q, −14q |
| 3 | PPTID WHO grade
II | Female | 24 years | – | – |
| 4 | Pineoblastoma WHO
grade IV | Male | 12 years | +12p, +12q,
+17p, +17q | −20p, −20q |
| 5 | Pineoblastoma WHO
grade IV | Female | 52 years | +8p, +8q, +20p,
+20q |
|
| 6 | Pineoblastoma WHO
grade IV | Female | 60 years | +1p, +9q,
+12q, +16p, +17p, +17q | -13q |
| 7 | Pineoblastoma WHO
grade IV | Male | 2
years | +8q, +9p,
+16p, +19p, +22q | −5q, −10q |
| 8 | Epidermoid
cyst | Male | 31 years | – | – |
| 9 | Germinoma | Male | 9
years | +1p, +9q,
+12q, +16p, +17p, +17q, +20q,
+22q | −3p, −7q, −9q |
| 10 | Germinoma | Male | 21 years | +1q,
+17q | −9p, -13q,
−15q |
| 11 | Germinoma | Male | 31 years | +7p,
+7q, +12q, +16p, +21q, +22q | −4p,
-13p |
| 12 | Germinoma | Male | 22 years | +1q, +7p,
+7q, +10p, +12p, +12q, +14q, +17q, +19p, +20q,
+21q | −1p, −3p, −3q, −4p,
−4q, −5p, −5q, −6p, −6q, −9p, −11p, −11q, -13q, −18q |
| 13 | Teratoma | Male | 5
years | +1p, +1q,
+7q, +8p, +8q, +9q, +11q, +12q, +16p,
+17p, +17q, +20q, +22q | −9p,
-13q |
| 14 | Pilocytic
astrocytoma WHO grade I | Male | 31 years | +1p, +1q,
+7q, +9q, +11q, +12q, +16p,
+17p, +17q, +20q, +22q |
|
| 15 | Pilocytic
astrocytoma WHO grade I | Male | 47 years | +7q,
+16p, +17p, +17q | −8q, −9p |
| 16 | Plexus
papilloma | Male | 5 months | +7p,
+7q, +12p, +12q, +14q, +15q, +19p, +21q |
|
| 17 | Solitary fibrous
tumor | Male | 35 years | +7p,
+7q, +9q, +16p, +17p, +17q,
+22q | -13q |
| 18 | Neuroendocrine
tumor | Male | 39 years | +5p, +5q,
+7p, +7q, +8p, +8q, +9p, +9q, +12p,
+12q, +21q, +22q | −1p, −1q, −2p, −2q,
−10p, −10q, -13q, −14q, −15q, −18p, −18q, −20p, −20q |
Comparative genomic hybridization
PPTs
The analyzed pineal cyst revealed gain of 16p as a
single chromosomal aberration. By contrast, one of the 2 grade II
PPTIDs did not reveal any chromosomal aberrations. In the other
grade II PPTID and in the 4 grade IV pineoblastomas, a mean of 8.2
net chromosomal aberrations (6.4 gains and 1.8 losses) was found.
Gains at 12q and 16p were observed in 3 out of 5 tumors. Moreover,
2 out of 4 pineoblastomas of WHO grade IV demonstrated gains of 8q
and whole chromosome 17, as well as loss of 13q (Table I).
Pineal GCTs
While the epidermoid cyst did not reveal any
chromosomal aberrations, the mature teratoma and 4 germinomas
analyzed showed a mean number of 12.8 net chromosomal changes, (8
gains and 4.8 losses). Common imbalances were gains of 12q and loss
of 13q in 3 out of 4 germinomas. Additionally, 4 germinomas showed
gains of 1q, 7, 16p, 17q and 22q, as well as losses of 9p. The
mature teratoma resembled similar chromosomal aberrations observed
in germinomas, also revealing gains of 1q, 7q, 16p, 17q and 22q,
and losses of 9p (Table I).
Glial tumors of the PR
Grade I pilocytic astrocytomas revealed gains of
11q, 7q, 16p, 20q and 22q, as well as gains of whole chromosome 1
and 17 (Table I).
Miscellaneous tumors of the PR
Plexus papilloma was characterized by gains of 7q,
12, 14q, 15q, 19p and 21q. No chromosomal losses were observed. SFT
of the PR showed gains on 7, 9q, 16p, 17 and 22q, and losses on
13q. The NET of the PR revealed the majority of cytogenetic net
changes of all analyzed tumors of the PR, including gains on 5, 7,
8, 9, 12p, 12q, 21q and 22q, and losses on 1, 2, 10, 13q, 14q, 15q,
18 and 20 (Table I).
Discussion
It can be expected that tumors originating from a
single histological cell population will display the same
cytogenetic pattern. However, classic concepts of tumorigenesis are
incomplete with regard to explaining the diversity of heterogenic
tumors of the PR (25). To date, the
cytogenetic and molecular data on PR tumors have been sparse. The
present study characterized 18 tumors of the PR and found no
distinctive, but rather common cytogenetic aberrations for each
tumor entity and origin.
Despite pineal cysts being considered as
non-neoplastic lesions, CGH analyses in the present study revealed
gains of 16p. The cytogenetic spectrum of grade II PPTID is
limited, revealing gains of 4q and 12q, and loss of chromosome 22
in two tumors each (1,26). One of the grade II PPTIDs in the
present study did not reveal any chromosomal imbalances, while the
other grade II PPTID, in addition to gains of 16p, showed a wide
range of gains and losses. By using CGH (1) and array-CGH (26,27), grade
IV pineoblastomas show relatively low chromosomal rearrangements,
with gains at 1, 12q, 13q and 19p, and losses at 13q, 16q and 22q
being the most frequent aberrations. In 2 out of 4 grade IV
pineoblastomas in the present study, gains at 16p could also be
observed. It may be concluded from the present results that gains
at 16p appear to be the chromosomal hallmark of PPTs, but no
correlation can be drawn between malignant progression and the
number of molecular cytogenetic changes.
Cytogenetic analysis of cerebral GCT, particularly
GCT of the PR, are seldom reported, with 27 pineal GCTs reported to
date (28,29). In these studies, the gain of 12p, a
hallmark in testicular GCT (30), was
detected at varying frequencies. The most common gains at 1q, 8q,
12p and 17q, as well as losses at 9q, 13q and 18q, have been
demonstrated in 27 GCTs of the PR, including the present study
cases (28,29). GCTs presented in the current study
commonly displayed gains at 1q (80%) and losses at 13q (60%). Gains
at 12p and 8q, and losses at 9q and 18q were observed in only 20%
of cases.
Notably, only WHO grade I pilocytic astrocytoma was
observed in the PR. Genetic analyses in grade I pilocytic
astrocytomas of different regions are scarce. Moreover, pilocytic
astrocytomas of different locations (hemispheric, cerebellar and
chiasmatic) appear to show different biological characteristics
with regard to biological aggressiveness (31). Gains have been described on
chromosomes 1p (63%), 2p (63%), 9p (63%), 9q (59.3%), 16p (63%),
17q, 19q (55.5%) and 22 (31,32). Losses have been shown for chromosomes
2p (40.7%), 3 (11%), 8p (63%), 9q, 12q, 16p (77.8%) and 16q
(31,32). It is not possible to determine a
correlation between these genetic alterations and the location of
the tumors. The present results showed gains on chromosomes 1p, 9q
and 16p, as well as additional chromosomal changes. Losses were
detected on chromosomes 8q and 9p. Losses for 2p, 3, 8p and 16p
were not detected. These results emphasize the heterogeneity of
grade I pilocytic astrocytomas I (31,33). Among
the miscellaneous tumors of the PR, a SFT, a plexus papilloma and
an NET were characterized. The present study is the first
cytogenetic characterization of a cerebral SFT occurring in the PR.
Chromosomal gains were observed for 7, 9q, 16p, 17 and 22q, and
loss at 13q. In 2010, the first study of a NET of the PR was
published (15). Until then, NET of
the PR had been unknown. The present study describes, for the first
time, the cytogenetic alterations observed in a NET of the PR. The
tumor demonstrated a high chromosomal instability with extended
gains and losses. Frequent chromosomal changes have been
demonstrated in NETs of the intestine, identifying several
chromosomal clusters and tumor groups stressing the complexity and
requirement for further CGH analysis of NETs of different locations
(34).
In summary, tumors of the PR in the present study,
regardless of histology and WHO grade, were characterized by
frequent gains at 7, 9q, 12q, 16p, 17 and 22q. Gain of 16p appeared
to be an common alteration in PPT. Gains at 12q were mainly
observed in PPT and GCT. Additionally, losses to 13q were
frequently observed in PPT and GCT, as well as in SFT and NET, but
not in grade I pilocytic astrocytomas of the PR. Therefore,
detection of chromosomal aberrations in these tumors may not be
indicative of histological differentiation. However, the present
data may of use for selecting genes of interest for higher
resolution analyses like array-CGH, or for more recent analyses
such as next generation sequencing (35).
Acknowledgements
Data shown in this publication are part of the
doctoral thesis of Florian Böhrnsen.
References
|
1
|
Rickert CH, Simon R, Bergmann M,
Dockhorn-Dworniczak B and Paulus W: Comparative genomic
hybridization in pineal parenchymal tumors. Genes Chromosomes
Cancer. 30:99–104. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Edwards MS, Hudgins RJ, Wilson CB, Levin
VA and Wara WM: Pineal region tumors in children. J Neurosurg.
68:689–697. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Regis J, Bouillot P, Rouby-Volot F,
Figarella-Branger D, Dufour H and Peragut JC: Pineal region tumors
and the role of stereotactic biopsy: Review of the mortality,
morbidity and diagnostic rates in 370 cases. Neurosurgery.
39:907–912. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Borja MJ, Plaza MJ, Altman N and Saigal G:
Conventional and advanced MRI features of pediatric intracranial
tumors: Supratentorial tumors. AJR Am J Roentgenol. 200:W483–W503.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeGirolami U and Schmidek H:
Clinicopathological study of 53 tumors of the pineal region. J
Neurosurg. 39:455–462. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang JK, Jeun SS, Hong YK, Park CK, Son
BC, Lee IW and Kim MC: Experience with pineal region tumors. Childs
Nerv Syst. 14:63–68. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Holou WN, Garton HJ, Muraszko KM,
Ibrahim M and Maher CO: Prevalence of pineal cysts in children and
young adults. Clinical article. J Neurosurg Pediatr. 4:230–236.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith AB, Rushing EJ and Smirniotopoulos
JG: From the archives of the AFIP: Lesions of the pineal region:
Radiologic-pathologic correlation. Radiographics. 30:2001–2020.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pettorini BL, Al-Mahfoud R, Jenkinson MD,
Avula S, Pizer B and Mallucci C: Surgical pathway and management of
pineal region tumours in children. Childs Nerv Syst. 2012.
|
|
11
|
Sajko T, Kudelic N, Lupret V, Lupret V Jr
and Nola IA: Treatment of pineal region lesions: Our experience in
39 patients. Coll Antropol. 33:1259–1263. 2009.PubMed/NCBI
|
|
12
|
Zhang J, Cheng H, Qiao Q, Zhang JS, Wang
YM, Fu X and Li Q: Malignant solitary fibrous tumor arising from
the pineal region: Case study and literature review.
Neuropathology. 30:294–298. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ozgural O, Kahilogullari G, Bozkurt M,
Heper AO and Savas A: Primary pineal glioblastoma: A case report.
Turk Neurosurg. 23:572–574. 2013.PubMed/NCBI
|
|
14
|
Carson BS, Weingart JD, Guarnieri M and
Fisher PG: Third ventricular choroid plexus papilloma with
psychosis. Case report. J Neurosurg. 87:103–105. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grozinsky-Glasberg S, Fichman S and Shimon
I: Metastatic bronchial neuroendocrine tumor to the pineal gland: A
unique manifestation of a rare disease. Hormones (Athens). 9:87–91.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korogi Y, Takahashi M and Ushio Y: MRI of
pineal region tumors. J Neurooncol. 54:251–261. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dumrongpisutikul N, Intrapiromkul J and
Yousem DM: Distinguishing between germinomas and pineal cell tumors
on MR imaging. AJNR Am J Neuroradiol. 33:550–555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tosi MR, Ricci R, Bottura G and Tugnoli V:
In vivo and in vitro nuclear magnetic resonance spectroscopy
investigation of an intracranial mass. Oncol Rep. 8:1337–1339.
2001.PubMed/NCBI
|
|
19
|
Legault G and Allen JC: Potential role of
ventricular tumor markers in CNS germ cell tumors. Pediatr Blood
Cancer. 60:1647–1650. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seregni E, Massimino M, Nerini Molteni S,
Pallotti F, van der Hiel B, Cefalo G, Spreafico F, Fossati F and
Bombardieri E: Serum and cerebrospinal fluid human chorionic
gonadotropin (hCG) and alpha-fetoprotein (AFP) in intracranial germ
cell tumors. Int J Biol Markers. 17:112–118. 2002.PubMed/NCBI
|
|
21
|
Leston J, Mottolese C, Champier J, Jouvet
A, Brun J, Sindou M, Chazot G, Claustrat B and Fèvre-Montange M:
Contribution of the daily melatonin profile to diagnosis of tumors
of the pineal region. J Neurooncol. 93:387–394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Korf HW, Gotz W, Herken R, Theuring F,
Gruss P and Schachenmayr W: S-antigen and rod-opsin immunoreactions
in midline brain neoplasms of transgenic mice: Similarities to
pineal cell tumors and certain medulloblastomas in man. J
Neuropathol Exp Neurol. 49:424–437. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Behari S, Jaiswal S, Nair P, Garg P and
Jaiswal AK: Tumors of the posterior third ventricular region in
pediatric patients: The Indian perspective and a review of
literature. J Pediatr Neurosci. 6:S56–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gutenberg A, Brandis A, Hong B, Gunawan B,
Enders C, Schaefer IM, Burger R, Ostertag H, Gaab M, Krauss JK, et
al: Common molecular cytogenetic pathway in papillary tumors of the
pineal region (PTPR). Brain Pathol. 21:672–677. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Berger AH, Knudson AG and Pandolfi PP: A
continuum model for tumour suppression. Nature. 476:163–169. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
von Bueren AO, Gerss J, Hagel C, Cai H,
Remke M, Hasselblatt M, Feuerstein BG, Pernet S, Delattre O,
Korshunov A, et al: DNA copy number alterations in central
primitive neuroectodermal tumors and tumors of the pineal region:
An international individual patient data meta-analysis. J
Neurooncol. 109:415–423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miller S, Rogers HA, Lyon P, Rand V,
Adamowicz-Brice M, Clifford SC, Hayden JT, Dyer S, Pfister S,
Korshunov A, et al: Genome-wide molecular characterization of
central nervous system primitive neuroectodermal tumor and
pineoblastoma. Neuro Oncol. 13:866–879. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schneider DT, Zahn S, Sievers S,
Alemazkour K, Reifenberger G, Wiestler OD, Calaminus G, Göbel U and
Perlman EJ: Molecular genetic analysis of central nervous system
germ cell tumors with comparative genomic hybridization. Mod
Pathol. 19:864–873. 2006.PubMed/NCBI
|
|
29
|
Rickert CH, Simon R, Bergmann M,
Dockhorn-Dworniczak B and Paulus W: Comparative genomic
hybridization in pineal germ cell tumors. J Neuropathol Exp Neurol.
59:815–821. 2000.PubMed/NCBI
|
|
30
|
Sheikine Y, Genega E, Melamed J, Lee P,
Reuter VE and Ye H: Molecular genetics of testicular germ cell
tumors. Am J Cancer Res. 2:153–167. 2012.PubMed/NCBI
|
|
31
|
Belirgen M, Berrak SG, Ozdag H, Bozkurt
SU, Eksioglu-Demiralp E and Ozek MM: Biologic tumor behavior in
pilocytic astrocytomas. Childs Nerv Syst. 28:375–389. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sanoudou D, Tingby O, Ferguson-Smith MA,
Collins VP and Coleman N: Analysis of pilocytic astrocytoma by
comparative genomic hybridization. Br J Cancer. 82:1218–1222.
2000.PubMed/NCBI
|
|
33
|
Lambert SR, Witt H, Hovestadt V, Zucknick
M, Kool M, Pearson DM, Korshunov A, Ryzhova M, Ichimura K, Jabado
N, et al: Differential expression and methylation of brain
developmental genes define location-specific subsets of pilocytic
astrocytoma. Acta Neuropathol. 126:291–301. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hashemi J, Fotouhi O, Sulaiman L, Kjellman
M, Höög A, Zedenius J and Larsson C: Copy number alterations in
small intestinal neuroendocrine tumors determined by array
comparative genomic hybridization. BMC Cancer. 13:5052013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Makedon F and Pearlman J: Tumor
classification based on DNA copy number aberrations determined
using SNP arrays. Oncol Rep. 15:1057–1059. 2006.PubMed/NCBI
|