Introduction
Colorectal cancer is one of the most common cancer
with leading cause of death (1). Its
classical molecular events have been well-studied. The oncogenes in
colorectal cancer are ras, scr and c-myc while the tumor suppressor
genes are APC and p53. The Wnt pathway is considered to be
important in the tumorgenesis of colorectal cancer. In 1990, Fearon
and Vogelstein (2) proposed a famous
model of colorectal cancer which believes a serials of gene and
signaling pathway alterations contribute to the histology changes
from normal tissue to adenoma and then to carcinoma. Li et
al found that at each stage of colorectal cancer, their gene
expression profiles were different (3). Jiang et al found that the early
stage colorectal cancer biomarkers and late stage biomarkers were
different and they can be connected by signal propagation on the
network (4). Many genes were found to
be associated with colorectal cancer by gene expression and network
analysis (5,6). And many signaling pathways, such as
Wnt/β-catenin signaling, epidermal growth factor receptor/Ras
signaling, p53 signaling, Notch signaling, Hedgehog signaling, and
Hippo signaling, were found to play roles in colorectal cancer
(7).
To summary the current understandings of colorectal
cancer, there are major mechanisms for colorectal cancer: (1) chromosome instability (CIN), (2) CpG island methylator phenotype (CIMP) and
(3) mismatch repair (MMR). In
approximately 85% of colorectal cancer patients, the chromosomal
instability (CIN) is observed (8).
They exhibited genomic instability on the chromosomal level. The
CIN patients usually have the poorest prognosis (9). In approximately 15–20% colorectal cancer
patients, there are widespread CIMP (10). In approximately 15% colorectal cancer
patients, Microsatellite instability (MSI) is detected (11). It is caused by the loss of DNA MMR
activity. The MSI patients tend to have a good prognosis (12). These mechanisms are not mutually
exclusive. For example, the MMR patients usually also show varying
degrees of CIN (8). Different
pathways that were used for characterizing each mechanism actually
can interact with each other and cross talk (7). Multiple signaling pathways share
transcription factors, microRNAs and ligases, such as miR-21,
miR-145, FBXW7 and β-TrCP (7).
To systematically investigate the relationship
between CIN, CIMP and MMR, we analyzed the gene expression profiles
of 585 colorectal cancer patients. These patients were annotated
with CIN, CIMP and MMR status. For each status, we applied advanced
minimal redundancy maximal relevance (mRMR) and incremental feature
selection (IFS) method to select its biomarkers genes. Then we
overlapped the CIN, CIMP and MMR biomarker genes. Since they may
not directly interact with each other, we used random walk with
restart (RWR) method to find the region that the CIN, CIMP and MMR
biomarker genes affect and investigated the commonly regulated
genes by CIN, CIMP and MMR. The biological functions of these
commonly regulated genes were analyzed. Our work found the
molecular cross talk among CIN, CIMP and MMR, revealed the internal
logic of colorectal tumorgenesis, and provided the emerging
therapeutic targets that may be suitable for most colorectal cancer
patients rather than a small proportion of patients.
Materials and methods
The gene expression profiles of 585
colorectal cancer patients
We downloaded the gene expression profiles of 585
colorectal cancer patients from GEO (Gene Expression Omnibus) with
accession number of GSE39582 (13).
The expression levels were measured with Affymetrix Human Genome
U133 Plus 2.0 Array which had 54,675 probes corresponding to 20,502
genes. The probes corresponding to the same gene were averaged. The
gene expression data was preprocessed with quantile normalization.
Within the 585 colon patients, there were 369 CIN+ and 112 CIN-, 93
CIMP+ and 420 CIMP-, 77 dMMR and 459 pMMR. For each analysis, the
patients with missing status were excluded. For example, for CIN+
and CIN-comparison, the 369 CIN+ and 112 CIN-patients were
considered while 104 without CIN information were excluded.
The CIN-associated gene selection
mRMR gene ranking
We used the mRMR method (14) to rank the genes based on their
relevance with CIN status and their redundancy between genes. The
mRMR method is based on information theory and has been widely used
in bioinformatics filed (15–19). To apply mRMR method, we used the C/C++
version mRMR software downloaded from http://home.penglab.com/proj/mRMR/. With mRMR method,
we obtained a ranked gene list. The top 500 mRMR genes were
analyzed.
IFS
To determine how many genes should be selected from
the mRMR gene list, we adopted the IFS method (4,20–24) and constructed 500 support vector
machine (SVM) classifiers. In this study, we used the svm function
with default parameters from R package e10171 (https://cran.r-project.org/web/packages/e1071/) to
build the SVM classifier. Each time, the top k genes in the mRMR
list was used to build the SVM classifier. And the performance of
the top k-gene classifier was evaluated with leave-one-out cross
validation (LOOCV). To objectively evaluate the classifier's
performance, Sensitivity (Sn), Specificity (Sp), Accuracy (ACC) and
Mathew's correlation coefficient (MCC) were calculated:
Sn=TPTP+FN
Sp=TNTN+FP
ACC=TP+TNTP+TN+FP+FN
MCC=TP×TN-FP×FN(TP+FP)(TP+FN)(TN+FP)(TN+FN)
where TP, TN, FP and FN stand for true positive
(CIN+), true negative (CIN-), false positive (CIN+) and false
negative (CIN-), respectively. Since the sizes of positive (CIN+)
and negative (CIN-) samples were imbalance in this study, MCC which
considered both Sn and Sp, was choose as the major measurement
(25). At last, based on the IFS
curve in which the number of top genes that were used as x-axis and
the LOOCV MCCs of classifiers as y-axis, we can decide how many
genes should be used to build a classifier with great performance
and small complexity. The peak or the change point of the IFS curve
were usually chosen.
The CIMP-associated gene
selection
Similarly, we can identify the CIMP-associated genes
using mRMR and IFS methods. Since the sample size of CIMP+ and
CIMP-patients were also imbalance, the MCC was considered as the
key measurement for prediction performance evaluation and was used
to plot the IFS curve.
The MMR-associated gene selection
Similarly, we can identify the MMR-associated genes
by analyzing the gene expression profiles pMMR and dMMR patients
using mRMR and IFS methods. The dMMR and pMMR were considered as
positive and negative samples, respectively. The MCC was used to
plot the IFS curve since there were much more pMMR than dMMR.
The overlapped genes and common
downstream genes of CIN, CIMP and MMR
We would like to known whether there is a general
mechanism for CIN, CIMP and MMR. The direct way is to overlap the
mRMR and IFS identified CIN associated genes, CIMP associated genes
and MMR associated genes.
Since the identified CIN associated genes, CIMP
associated genes and MMR associated genes may be incomplete or
locate at the upstream of the colorectal cancer signaling pathway,
we tried to pin down the area affected by the CIN associated genes,
CIMP associated genes and MMR associated genes on the
protein-protein interaction network of using RWR method (26–29). The
STRING network (version 10.0) (30)
is a comprehensive protein-protein functional association network
that has been widely used (26,28,31–39).
It included 19,247 proteins and 4,274,001 interactions. We
constructed the network using the protein-protein interactions with
confidence score >0.900 which is the highest confidence
interaction in STRING database. Then the n*n adjacent matrix
(A) of the network which included n proteins was column-wise
normalized to make the column sum to be 1 by assign 1/m to the m
interaction proteins of protein j in column j and 0 to other
proteins without interactions.
The random walk procedure repeat in every time tick
(t→t+1) from the initial seed genes which were represented as a n
length vector with P0 value of 1/k for the k seed
genes and value of 0 for other n-k non-seed genes. The state
probabilities Pt+1 at time t+1 is calculated as
follow:
Pt+1=(1-r)APt+rP0
(5), where Pt is
state probabilities at time t, r is the restart probability which
is set to 0.7 as suggested by previous studies (26–29,40). It
has been reported that if r is in a sizable range (0.5–0.8), the
results will have little difference (40). These random walk process will stop
when the difference between two steps is smaller than 1e-6. At
last, all genes on the network will be assigned with a RWR score
which corresponds to the probability of being expanded from the
seed genes.
To statically evaluate the significance of RWR
score, we randomly chosen the same number of seed genes and
calculated their RWR scores for 1,000 times. The significance of
actual RWR score can be defined as a permutation P-value of how
times the random RWR scores was greater than the actual RWR score
over the permutation times which was 1,000 in this study. The genes
with permutation P-value smaller than 0.05 were considered as
significant RWR expanded genes.
The RWR expanded genes can represent the downstream
genes of CIN, CIMP and MMR and be used for common downstream gene
analysis. The functions of the common CIN, CIMP and MMR downstream
genes were enriched onto KEGG pathways and Gene Ontology (GO) terms
using hypergeometric test.
Results and Discussion
The CIN associated genes identified
with mRMR and IFS
The top 500 most discriminative genes between CIN+
and CIN-samples were ranked using the mRMR method which considered
both their relevance with CIN status, and their redundancy with
selected genes. After the genes were ranked by mRMR, we chosen the
number of top genes by applying the IFS procedure. Different number
of top genes were tried and their prediction performance were
evaluated. The IFS curve with the number of genes as x-axis and
leave one out cross validation MCC as y-axis was shown in Fig. 1A. It can be seen that when 34 genes
were used, the leave one out cross validation MCC was the highest.
The leave one out cross validation Sn, Sp, ACC and MCC of these 34
genes were 0.932, 0.696, 0.877 and 0.648, respectively. Therefore
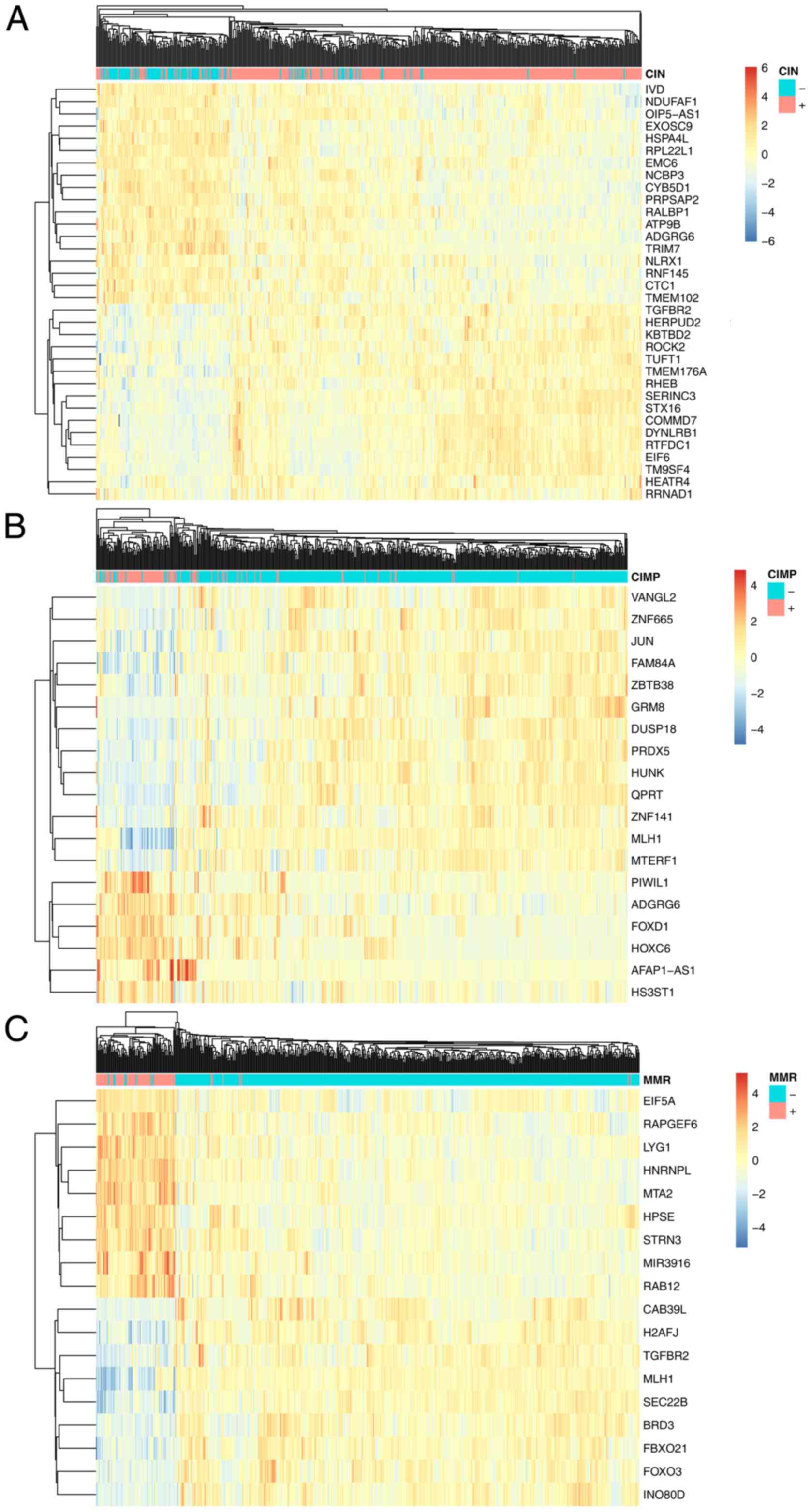
these 34 genes were chosen and shown in Table I. As shown in Fig. 2A, the 34 CIN associated genes can
cluster the CIN+ and CIN-patients into the right groups. IVD,
NDUFAF1, OIP5-AS1, EXOSC9, HSPA4L, RPL22L1, EMC6, NCBP3, CYB5D1,
PRPSAP2, RALBP1, ATP9B, ADGRG6, TRIM7, NLRX1, RNF145, CTC1, TMEM102
were highly expressed in CIN-patients while TGFBR2, HERPUD2,
KBTBD2, ROCK2, TUFT1, TMEM176A, RHEB, SERINC3, STX16, COMMD7,
DYNLRB1, RTFDC1, EIF6, TM9SF4, HEATR4, RRNAD1 were highly expressed
in CIN+ patients.
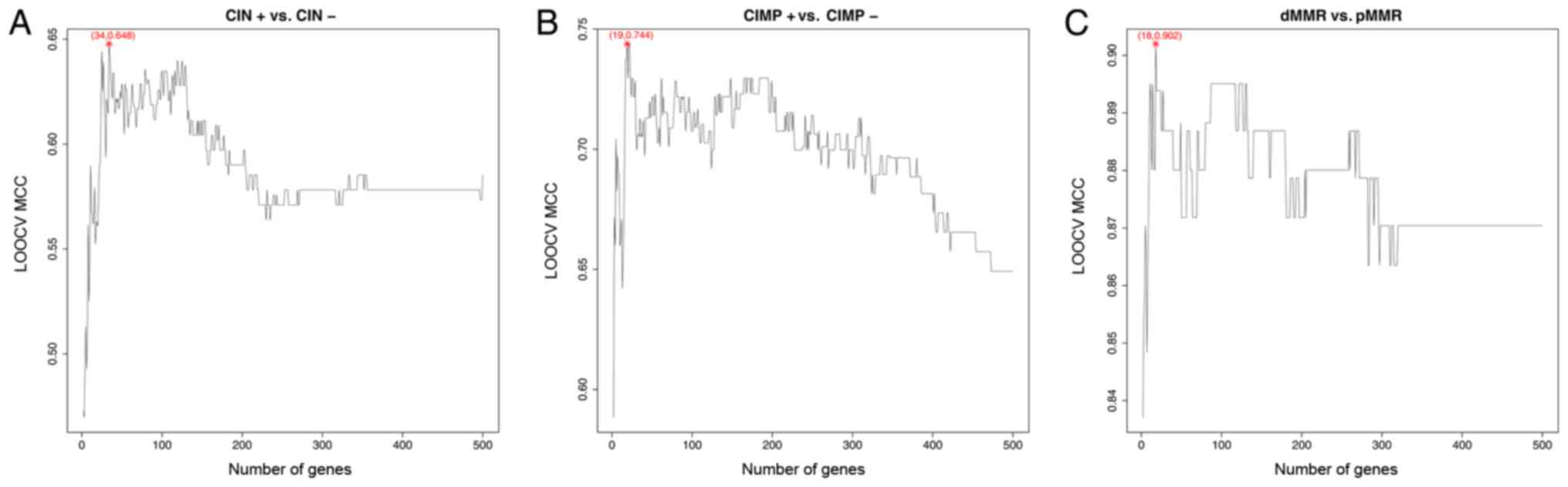 | Figure 1.The IFS curves of CIN, CIMP and MMR.
(A) The IFS curve of CIN. The top 34 mRMR genes were chosen and
their LOOCV sensitivity, specificity, accuracy and MCC were 0.932,
0.696, 0.877 and 0.648, respectively. (B) The IFS curve of CIMP.
The top 19 mRMR genes were chosen and their leave one out cross
validation sensitivity, specificity, accuracy and MCC were 0.710,
0.976, 0.928 and 0.744, respectively. (C) The IFS curve of MMR. The
top 18 mRMR genes were chosen and their leave one out cross
validation sensitivity, specificity, accuracy and MCC were 0.922,
0.985, 0.976 and 0.902, respectively. IFS, incremental feature
selection; CIN, chromosome instability; CIMP, CpG island methylator
phenotype; MMR, mismatch repair; mRMR, minimal redundancy maximal
relevance; LOOCV, leave-one-out cross validation. |
 | Table I.The 34 chromosome
instability-associated genes. |
Table I.
The 34 chromosome
instability-associated genes.
| Order | Symbol | Name | Entrez gene | mRMR score |
|---|
| 1 | STX16 | Syntaxin 16 | 8675 | 0.161 |
| 2 | NCBP3 | Nuclear cap binding
subunit 3 | 55421 | 0.062 |
| 3 | IVD | Isovaleryl-CoA
dehydrogenase | 3712 | 0.061 |
| 4 | DYNLRB1 | Dynein light chain
roadblock-type 1 | 83658 | 0.067 |
| 5 | EXOSC9 | Exosome component
9 | 5393 | 0.044 |
| 6 | ATP9B | ATPase phospholipid
transporting 9B (putative) | 374868 | 0.043 |
| 7 | KBTBD2 | Kelch repeat and
BTB domain containing 2 | 25948 | 0.042 |
| 8 | EMC6 | ER membrane protein
complex subunit 6 | 83460 | 0.043 |
| 9 | ADGRG6 | Adhesion G
protein-coupled receptor G6 | 57211 | 0.046 |
| 10 | OIP5-AS1 | OIP5 antisense RNA
1 | 729082 | 0.044 |
| 11 | RNF145 | Ring finger protein
145 | 153830 | 0.043 |
| 12 | COMMD7 | COMM domain
containing 7 | 149951 | 0.046 |
| 13 | TUFT1 | Tuftelin 1 | 7286 | 0.038 |
| 14 | NLRX1 | NLR family member
X1 | 79671 | 0.036 |
| 15 | CYB5D1 | Cytochrome b5
domain containing 1 | 124637 | 0.038 |
| 16 | RTFDC1 | Replication
termination factor 2 domain containing 1 | 51507 | 0.037 |
| 17 | RPL22L1 | Ribosomal protein
L22 like 1 | 200916 | 0.034 |
| 18 | TMEM102 | Transmembrane
protein 102 | 284114 | 0.032 |
| 19 | TM9SF4 | Transmembrane 9
superfamily member 4 | 9777 | 0.035 |
| 20 | HERPUD2 | HERPUD family
member 2 | 64224 | 0.033 |
| 21 | RHEB | Ras homolog
enriched in brain | 6009 | 0.033 |
| 22 | NDUFAF1 | NADH:ubiquinone
oxidoreductase complex assembly factor 1 | 51103 | 0.033 |
| 23 | TGFBR2 | Transforming growth
factor β receptor 2 | 7048 | 0.034 |
| 24 | TRIM7 | Tripartite motif
containing 7 | 81786 | 0.032 |
| 25 | PRPSAP2 | Phosphoribosyl
pyrophosphate synthetase associated protein 2 | 5636 | 0.032 |
| 26 | HEATR4 | HEAT repeat
containing 4 | 399671 | 0.032 |
| 27 | SERINC3 | Serine incorporator
3 | 10955 | 0.034 |
| 28 | HSPA4L | Heat shock protein
family A (Hsp70) member 4 like | 22824 | 0.03 |
| 29 | RALBP1 | RalA binding
protein 1 | 10928 | 0.029 |
| 30 | RRNAD1 | Ribosomal RNA
adenine dimethylase domain containing 1 | 51093 | 0.029 |
| 31 | CTC1 | CST telomere
replication complex component 1 | 80169 | 0.03 |
| 32 | EIF6 | Eukaryotic
translation initiation factor 6 | 3692 | 0.031 |
| 33 | TMEM176A | Transmembrane
protein 176A | 55365 | 0.031 |
| 34 | ROCK2 | Rho associated
coiled-coil containing protein kinase 2 | 9475 | 0.03 |
The CIMP associated genes identified
with mRMR and IFS
Similarly, the CIMP associated genes can be
identified using mRMR and IFS methods. As a result, 19 genes were
selected based on the IFS curve shown in Fig. 1B and listed in Table II. The 19 genes' leave one out cross
validation Sn, Sp, ACC and MCC were 0.710, 0.976, 0.928 and 0.744,
respectively. As shown in Fig. 2B,
the 19 CIMP associated genes can cluster the CIMP+ and
CIMP-patients into the right groups. VANGL2, ZNF665, JUN, FAM84A,
ZBTB38, GRM8, DUSP18, PRDX5, HUNK, QPRT, ZNF141, MLH1, MTERF1 were
highly expressed in CIMP-patients while PIWIL1, ADGRG6, FOXD1,
HOXC6, AFAP1-AS1, HS3ST1 were highly expressed in CIMP+
patients.
| Table II.The 19 CpG island methylator
phenotype-associated genes. |
Table II.
The 19 CpG island methylator
phenotype-associated genes.
| Order | Name | Gene name | Entrez gene | mRMR score |
|---|
| 1 | MLH1 | mutL homolog 1 | 4292 | 0.193 |
| 2 | HUNK | Hormonally
up-regulated Neu-associated kinase | 30811 | 0.069 |
| 3 | ZNF141 | Zinc finger protein
141 | 7700 | 0.063 |
| 4 | DUSP18 | Dual specificity
phosphatase 18 | 150290 | 0.058 |
| 5 | ADGRG6 | Adhesion G
protein-coupled receptor G6 | 57211 | 0.053 |
| 6 | FOXD1 | Forkhead box
D1 | 2297 | 0.052 |
| 7 | FAM84A | Family with
sequence similarity 84 member A | 151354 | 0.049 |
| 8 | AFAP1-AS1 | AFAP1 antisense RNA
1 | 84740 | 0.047 |
| 9 | ZBTB38 | Zinc finger and BTB
domain containing 38 | 253461 | 0.052 |
| 10 | VANGL2 | VANGL planar cell
polarity protein 2 | 57216 | 0.054 |
| 11 | PRDX5 | Peroxiredoxin
5 | 25824 | 0.049 |
| 12 | MTERF1 | Mitochondrial
transcription termination factor 1 | 7978 | 0.05 |
| 13 | QPRT | Quinolinate
phosphoribosyltransferase | 23475 | 0.05 |
| 14 | HOXC6 | Homeobox C6 | 3223 | 0.045 |
| 15 | HS3ST1 | Heparan
sulfate-glucosamine 3-sulfotransferase 1 | 9957 | 0.044 |
| 16 | PIWIL1 | Piwi like
RNA-mediated gene silencing 1 | 9271 | 0.046 |
| 17 | JUN | Jun proto-oncogene,
AP-1 transcription factor subunit | 3725 | 0.047 |
| 18 | GRM8 | Glutamate
metabotropic receptor 8 | 2918 | 0.045 |
| 19 | ZNF665 | Zinc finger protein
665 | 79788 | 0.046 |
The MMR associated genes identified
with mRMR and IFS
Similarly, the MMR associated genes can be
identified using mRMR and IFS methods. As a result, 18 genes were
selected based on the IFS curve shown in Fig. 1C and listed in Table III. The leave one out cross
validation Sn, Sp, ACC and MCC of these 18 genes were 0.922, 0.985,
0.976 and 0.902, respectively. As shown in Fig. 2C, the 18 MMR associated genes can
cluster the MMR+ and MMR-patients into the right groups. CAB39L,
H2AFJ, TGFBR2, MLH1, SEC22B, BRD3, FBXO21, FOXO3, INO80D were
highly expressed in MMR-patients while EIF5A, RAPGEF6, LYG1,
HNRNPL, MTA2, HPSE, STRN3, MIR3916, RAB12 were highly expressed in
MMR+ patients.
| Table III.The 18 mismatch repair-associated
genes. |
Table III.
The 18 mismatch repair-associated
genes.
| Order | Name | Gene name | Entrez gene | mRMR score |
|---|
| 1 | HNRNPL | Heterogeneous
nuclear ribonucleoprotein L | 3191 | 0.285 |
| 2 | HPSE | Heparanase | 10855 | 0.097 |
| 3 | CAB39L | Calcium binding
protein 39 like | 81617 | 0.081 |
| 4 | MTA2 | Metastasis
associated 1 family member 2 | 9219 | 0.093 |
| 5 | RAPGEF6 | Rap guanine
nucleotide exchange factor 6 | 51735 | 0.086 |
| 6 | LYG1 | Lysozyme g1 | 129530 | 0.081 |
| 7 | SEC22B | SEC22 homolog B,
vesicle trafficking protein (gene/pseudogene) | 9554 | 0.081 |
| 8 | BRD3 | Bromodomain
containing 3 | 8019 | 0.076 |
| 9 | H2AFJ | H2A histone family
member J | 55766 | 0.079 |
| 10 | RAB12 | RAB12, member RAS
oncogene family | 201475 | 0.072 |
| 11 | TGFBR2 | Transforming growth
factor β receptor 2 | 7048 | 0.078 |
| 12 | STRN3 | Striatin 3 | 29966 | 0.076 |
| 13 | INO80D | INO80 complex
subunit D | 54891 | 0.076 |
| 14 | MLH1 | MutL homolog 1 | 4292 | 0.079 |
| 15 | EIF5A | Eukaryotic
translation initiation factor 5A | 1984 | 0.072 |
| 16 | MIR3916 | microRNA 3916 | 100500849 | 0.069 |
| 17 | FOXO3 | Forkhead box
O3 | 2309 | 0.069 |
| 18 | FBXO21 | F-box protein
21 | 23014 | 0.069 |
The direct overlap between CIN
associated genes, CIMP associated genes and MMR associated
genes
As three major mechanisms of colorectal cancer, we
would like to investigate whether there were overlaps between CIN
associated genes, CIMP associated genes and MMR associated genes.
The Venn diagram of CIN associated genes, CIMP associated genes and
MMR associated genes were shown in Fig.
3. It can be seen that none genes were common in these three
gene lists. The overlap between CIN and CIMP was ADGRG6, the common
gene between CIN and MMR was TGFBR2 and the overlap between CIMP
and MMR was MLH1. The references of ADGRG6 was limited and its
functions were largely unknown. Interestingly, TGFBR2 has been
reported as a candidate driver gene in MSI colorectal cancer
(41) and the MMR patients usually
also show varying degrees of CIN (8).
TGFBR2 may be key of the association of CIN and MMR. The
correlation of MLH1 methylation and MMR status has been reported
(42) and it confirmed the
association of CIMP and MMR.
The cross talk between CIN, CIMP and
MMR
Since there is little overlap between the CIN
associated genes, CIMP associated genes and MMR associated genes
identified by mRMR and IFS, we would like to investigate whether
they have common downstream genes. To verify this, we used the
workflow shown in Fig. 4 to
investigate the cross talk between CIN, CIMP and MMR. The key is
step (C) which identifies the genes that the CIN, CIMP and MMR
affects, i.e. the downstream genes of CIN, CIMP and MMR. To do so,
first we mapped the CIN associated genes onto the network and then,
expanded them using RWR network on the network. At last, by
comparing with random permutations, the significant RWR expanded
genes were identified as the downstream of CIN. Similarly, the
downstream genes of CIMP and MMR can be identified.
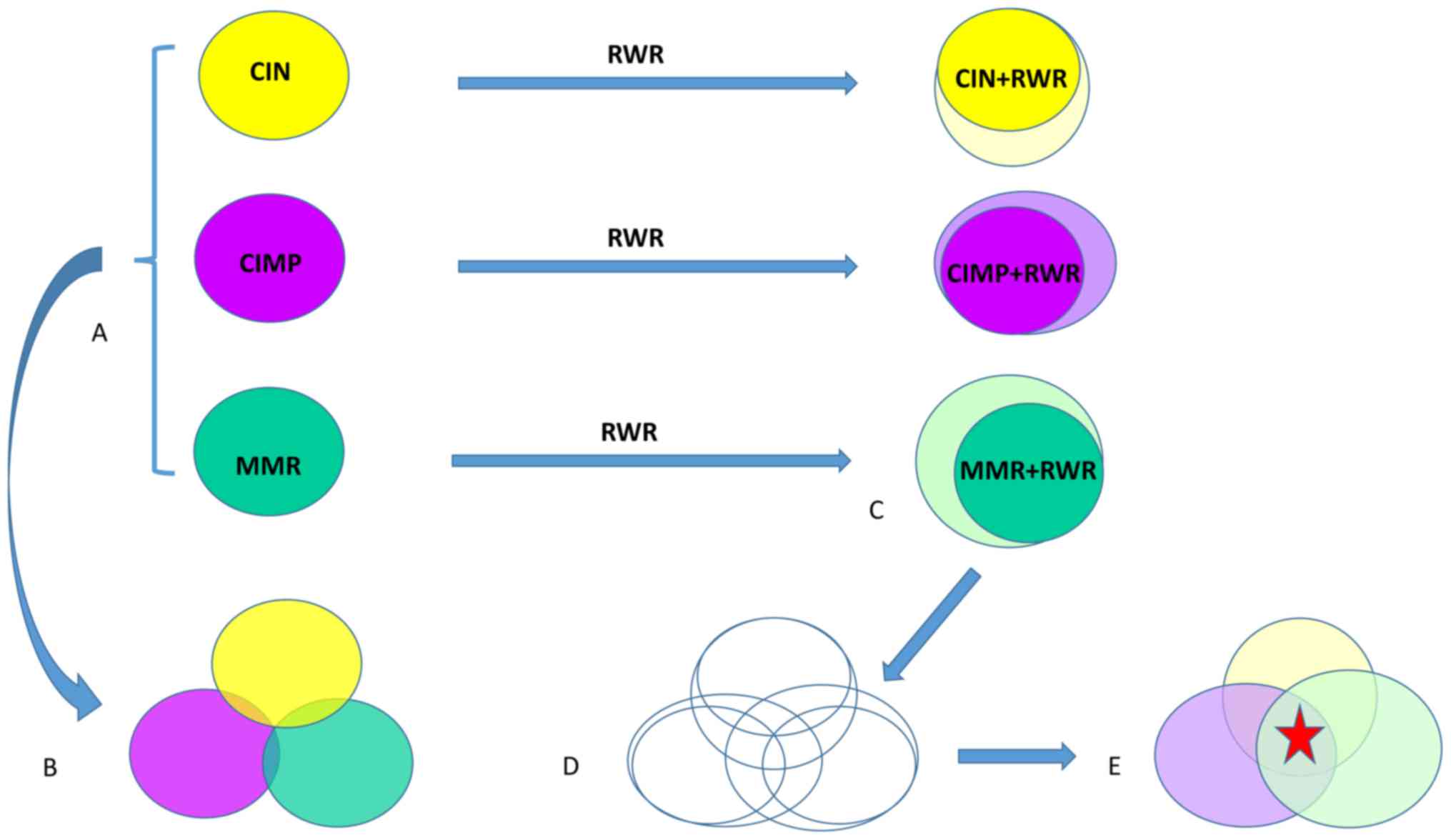 | Figure 4.The workflow to investigate the cross
talk among CIN, CIMP and MMR. (A) The CIN associated genes, CIMP
associated genes and MMR associated genes were identified using
mRMR and IFS methods. (B) The direct overlap between CIN genes,
CIMP genes and MMR genes were little. (C) The genes that the CIN
genes, CIMP genes and MMR genes affect were identified using RWR
method. (D) When both the CIN genes, CIMP genes and MMR genes and
their RWR genes were considered, the overlap among CIN, CIMP and
MMR was significantly increased. (E) The biological functions of
the common genes (the red star) were studied. CIN, chromosome
instability; CIMP, CpG island methylator phenotype; MMR, mismatch
repair; mRMR, minimal redundancy maximal relevance; IFS,
incremental feature selection; RWR, random walk with restart. |
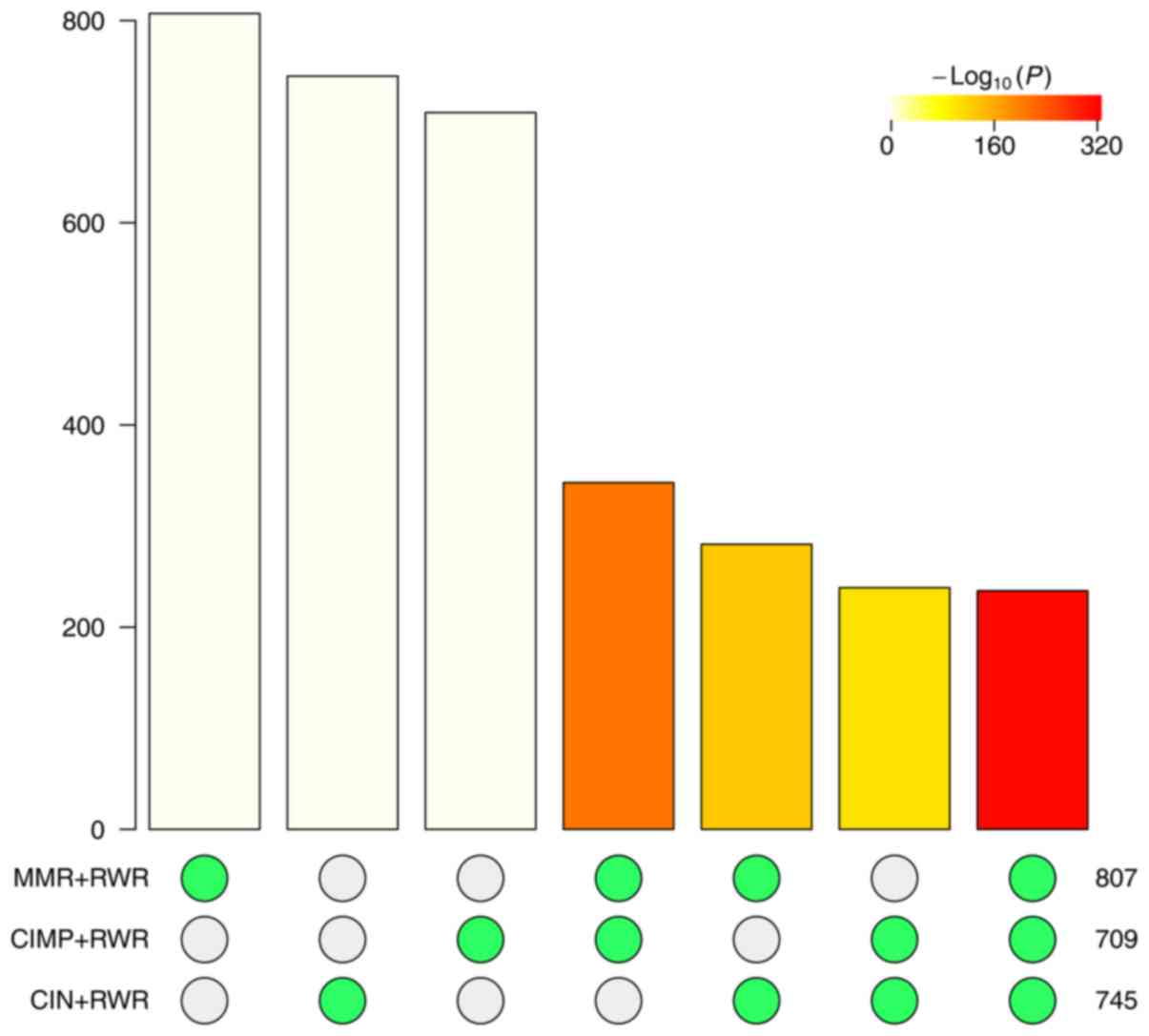
The numbers of downstream genes of CIN, CIMP and MMR
with permutation P-value <0.05 were 745, 709 and 807,
respectively. Fig. 5 showed the
overlap among CIN, CIMP and MMR and there were 236 common
downstream genes of CIN, CIMP and MMR. These 236 genes were shown
in Table IV. To statistically
evaluate the significance of overlap, we calculated the odds ratio
and P-value using R package Super Exact Test (43). The results were shown in Fig. 6. The odds ratio of overlap was 60.3
and the P-value was smaller than 1e-320.
| Table IV.Common downstream genes of chromosome
instability, CpG island methylator phenotype and mismatch
repair. |
Table IV.
Common downstream genes of chromosome
instability, CpG island methylator phenotype and mismatch
repair.
| List of common
genes |
|---|
| A1BG | CD248 | DEFB131 | HECA | LCE1A | NAIP | SEMA4C | TRMU |
| A1CF | CDKAL1 | DEFB134 | HES2 | LCE1B | NCDN | SERINC3 | TSEN15 |
| ABCC5 | CEP120 | DEFB135 | HES3 | LCE1D | NCR3 | SERINC5 | TSEN2 |
| ABHD12 | CFAP58 | DNAJC9 | HGFAC | LCE1E | NCR3LG1 | SETDB2 | TSEN34 |
| ABHD6 | CGREF1 | DYSF | HHLA2 | LCE3B | NSUN4 | SLC16A7 | TSEN54 |
| ABI3 | CGRRF1 | EMB | HHLA3 | LCE3C | NTNG1 | SLC30A8 | UBAP2 |
| ABI3BP | CLASRP | ENAM | HLA-DOA | LCN1 | NTNG2 | SLC36A2 | UNKL |
| ACOT13 | CLEC2A | ETV7 | HLA-DOB | LETM1 | OR10H1 | SLC3A1 | UPK1A |
| ADAT1 | CLK2 | FAM149B1 | HMGN3 | LIAS | ORAOV1 | SLC51A | UPK1B |
| ADAT2 | CLK3 | FAM3C | HOXC13 | LIPT2 | PANK1 | SLC51B | UPK2 |
| ADAT3 | CLK4 | FAT4 | HPCA | LMBR1L | PANK2 | SLC6A18 | UPK3A |
| AGR2 | CLN6 | FBXO38 | IGLL1 | LRRC4 | PANK3 | SLC6A19 | UPK3B |
| AGR3 | CLN8 | FJX1 | IGSF3 | LRRC4C | PANK4 | SLC6A20 | VEZT |
| AMBN | CNBD1 | FLCN | IGSF6 | LXN | PCTP | SLC6A9 | VN1R1 |
| AMICA1 | COASY | FNIP2 | IGSF9B | LYPD3 | PHF11 | SLC7A9 | VNN2 |
| ANO5 | COMMD10 | FOXQ1 | IKBIP | MARCO | PIP | SMDT1 | VPREB1 |
| APOBEC1 | COMMD7 | FUZ | INTU | MCU | PLXDC1 | SP8 | XAGE1B |
| AZGP1 | COMMD8 | GABRR1 | KBTBD6 | MDGA1 | PPCDC | SPICE1 | XAGE2 |
| BCS1L | CPA1 | GABRR2 | KBTBD7 | METTL9 | PPCS | SPINK9 | YAE1D1 |
| BFSP1 | CPA4 | GNPTAB | KCNK10 | MFSD10 | PRLH | SPINT1 | YIPF3 |
| BFSP2 | CPN1 | GNPTG | KCNK2 | MICU1 | PRLHR | ST14 | YIPF4 |
| BSCL2 | CPN2 | GP2 | KCNK4 | MICU2 | PRSS8 | STYX | YRDC |
| CARHSP1 | CRISP3 | GRID2 | KIAA0319 | MMS22L | PTCD3 | SUGP2 | ZCCHC17 |
| CCDC109B | CRYBA1 | GRID2IP | KLF7 | MSRA | RASD2 | TM2D1 | ZFR |
| CCDC179 | CRYBB1 | GRXCR1 | KLK5 | MSRB2 | RBBP9 | TM2D2 | ZNF461 |
| CCDC68 | CTAGE5 | GSX2 | KLRF2 | MSRB3 | RPUSD4 | TMEM126B | ZNF772 |
| CCIN | CXADR | GTPBP1 | KRTAP24-1 | MTERF4 | RSRP1 | TMEM19 |
|
| CD101 | CYLC1 | GTPBP3 | KRTAP25-1 | MTO1 | SCGB2A2 | TMEM27 |
|
| CD200 | DCDC2 | HAS1 | KRTAP27-1 | MUCL1 | SCGB3A2 | TONSL |
|
| CD200R1 | DEFB110 | HAS3 | L3MBTL1 | MYO7A | SDCBP2 | TOX2 |
|
The biological functions of the overlapped genes
were investigated by enriching them onto KEGG and GO. The
enrichment results were summarized in Table V. It can be seen that the
significantly enriched KEGG pathways with FDR (false discovery
rate) <0.05 were: hsa00770 Pantothenate and CoA biosynthesis,
hsa00785 Lipoic acid metabolism and hsa04514 Cell adhesion
molecules (CAMs). Similarly, the most significantly enriched GO
terms were: GO:0015937 coenzyme A biosynthetic process, GO:0015936
coenzyme A metabolic process, GO:0033866 nucleoside bisphosphate
biosynthetic process, GO:0034030_ribonucleoside bisphosphate
biosynthetic process and GO:0034033 purine nucleoside bisphosphate
biosynthetic process. These results indicated that the CIN, CIMP
and MMR all affect biosynthetic and metabolic process and pathway
to accelerate the tumorgenesis. In clinic, the metabolic syndrome
was found to be able to increase the risk of colorectal cancer
(44). And in colorectal cancer cell,
there are aberration of various metabolites, such as nucleotides,
amino acids, tricarboxylic acid, carbohydrates, and
pentose-phosphate (45).
| Table V.Kyoto Encyclopedia of Genes and
Genomes and Gene Ontology enrichments of common downstream genes of
chromosome instability, CpG island methylator phenotype and
mismatch repair. |
Table V.
Kyoto Encyclopedia of Genes and
Genomes and Gene Ontology enrichments of common downstream genes of
chromosome instability, CpG island methylator phenotype and
mismatch repair.
| Type | Gene set | FDR |
|---|
| KEGG | hsa00770
Pantothenate and CoA biosynthesis | 4.35E-11 |
|
| hsa00785 Lipoic
acid metabolism | 0.0226 |
|
| hsa04514 Cell
adhesion molecules (CAMs) | 0.0476 |
| GO BP | GO:0015937 coenzyme
A biosynthetic process | 9.66E-08 |
|
| GO:0015936 coenzyme
A metabolic process | 1.32E-06 |
|
| GO:0033866
nucleoside bisphosphate biosynthetic process | 1.72E-06 |
|
| GO:0034030
ribonucleoside bisphosphate biosynthetic process | 1.72E-06 |
|
| GO:0034033 purine
nucleoside bisphosphate biosynthetic process | 1.72E-06 |
|
| GO:0008033 tRNA
processing | 6.32E-05 |
|
| GO:0009451 RNA
modification | 0.000240 |
|
| GO:0033865
nucleoside bisphosphate metabolic process | 0.000267 |
|
| GO:0033875
ribonucleoside bisphosphate metabolic process | 0.000267 |
|
| GO:0034032 purine
nucleoside bisphosphate metabolic process | 0.000267 |
|
| GO:0015804 neutral
amino acid transport | 0.000561 |
|
| GO:0006865 amino
acid transport | 0.00215 |
|
| GO:0015807 L-amino
acid transport | 0.00215 |
|
| GO:0046942
carboxylic acid transport | 0.00218 |
|
| GO:0000379
tRNA-type intron splice site recognition and cleavage | 0.00218 |
|
| GO:0006399 tRNA
metabolic process | 0.00233 |
|
| GO:0015849 organic
acid transport | 0.00254 |
|
| GO:0036444
mitochondrial calcium uptake | 0.00277 |
|
| GO:0015711 organic
anion transport | 0.00408 |
|
| GO:0008544
epidermis development | 0.00408 |
|
| GO:0031424
keratinization | 0.00458 |
|
| GO:0030855
epithelial cell differentiation | 0.00458 |
|
| GO:0006820 anion
transport | 0.00458 |
|
| GO:1905039
carboxylic acid transmembrane transport | 0.00473 |
| GO MF | GO:0000213
tRNA-intron endonuclease activity | 5.22E-05 |
|
| GO:0004594
pantothenate kinase activity | 5.22E-05 |
|
| GO:0015171 amino
acid transmembrane transporter activity | 0.000748 |
|
| GO:0008514 organic
anion transmembrane transporter activity | 0.000748 |
|
| GO:0046943
carboxylic acid transmembrane transporter activity | 0.000760 |
|
| GO:0008509 anion
transmembrane transporter activity | 0.00112 |
|
| GO:0005342 organic
acid transmembrane transporter activity | 0.00112 |
|
| GO:0015175 neutral
amino acid transmembrane transporter activity | 0.00162 |
|
| GO:0016892
endoribonuclease activity, producing 3′-phosphomonoesters | 0.00230 |
|
| GO:0004549
tRNA-specific ribonuclease activity | 0.0128 |
|
| GO:0015179 L-amino
acid transmembrane transporter activity | 0.0221 |
|
| GO:0005328
neurotransmitter:sodium symporter activity | 0.0291 |
|
| GO:0005212
structural constituent of eye lens | 0.0333 |
|
| GO:0016894
endonuclease activity, active with either ribo- or deoxyribonucleic
acids and producing 3′-phosphomonoesters | 0.0458 |
|
| GO:0008251
tRNA-specific adenosine deaminase activity | 0.0462 |
| GO CC | GO:1990246 uniplex
complex | 0.000199 |
|
| GO:0000214
tRNA-intron endonuclease complex | 0.00661 |
|
| GO:0005886 plasma
membrane | 0.0114 |
|
| GO:0071944 cell
periphery | 0.0114 |
|
| GO:0031526 brush
border membrane | 0.0125 |
|
| GO:0098590 plasma
membrane region | 0.0125 |
|
| GO:0098862 cluster
of actin-based cell projections | 0.0148 |
|
| GO:0044459 plasma
membrane part | 0.0168 |
|
| GO:0001533
cornified envelope | 0.0242 |
As a complex disease, the colorectal cancer can be
caused by several different mechanisms. The three well-known one
were CIN, CIMP and MMR. They were different but not exclusive. We
investigated the genes that were associated with CIN, CIMP and MMR,
separately using mRMR and IFS methods. Then by direct overlapping
the CIN associated genes, CIMP associated genes and MMR associated
genes, they share little common genes. Therefore, they were highly
possible to interact with each other indirectly. To verify this
idea, we identified the downstream genes that the CIN associated
genes, CIMP associated genes and MMR associated genes may affect
using RWR method. After the RWR analysis, the overlap between CIN,
CIMP and MMR become significantly greater and the common downstream
genes were involved in biosynthetic and metabolic process and
pathway. These results can help explain the non-exclusiveness of
CIN, CIMP and MMR and why they may co-occur from a protein-protein
interaction network view. What's more, the common genes of CIN,
CIMP and MMR can be possible targets of new broad-spectrum
anti-cancer drugs that can treat more patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by Health and Family
Planning Commission of Zhejiang Province (grant no. 2013kYA212),
National Natural Science Foundation of China (grant no. 31701151),
Shanghai Sailing Program and The Youth Innovation Promotion
Association of Chinese Academy of Sciences (CAS) (grant no.
2016245).
Availability of data and materials
The gene expression profiles of 585 colorectal
cancer patients were obtained from GEO (Gene Expression Omnibus)
with accession number of GSE39582.
Authors' contributions
RFW and TH designed the experiment. TMZ and TH
performed the experiment, analyzed the data and wrote the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li BQ, Huang T, Zhang J, Zhang N, Huang
GH, Liu L and Cai YD: An ensemble prognostic model for colorectal
cancer. PLoS One. 8:e634942013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Huang T, Chen L, Gao YF, Cai Y
and Chou KC: Signal propagation in protein interaction network
during colorectal cancer progression. BioMed Research
International. 2013:2870192013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li BQ, Huang T, Liu L, Cai YD and Chou KC:
Identification of colorectal cancer related genes with mRMR and
shortest path in protein-protein interaction network. PLoS One.
7:e333932012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang T, Li BQ and Cai YD: The integrative
network of gene expression, microRNA, methylation and copy number
variation in colon and rectal cancer. Curr Bioinform. 11:59–65.
2016. View Article : Google Scholar
|
|
7
|
Wu WK, Wang XJ, Cheng AS, Luo MX, Ng SS,
To KF, Chan FK, Cho CH, Sung JJ and Yu J: Dysregulation and
crosstalk of cellular signaling pathways in colon carcinogenesis.
Crit Rev Oncol Hematol. 86:251–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trautmann K, Terdiman JP, French AJ,
Roydasgupta R, Sein N, Kakar S, Fridlyand J, Snijders AM, Albertson
DG, Thibodeau SN and Waldman FM: Chromosomal instability in
microsatellite-unstable and stable colon cancer. Clin Cancer Res.
12:6379–6385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walther A, Houlston R and Tomlinson I:
Association between chromosomal instability and prognosis in
colorectal cancer: A meta-analysis. Gut. 57:941–950. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vedeld HM, Merok M, Jeanmougin M,
Danielsen SA, Honne H, Presthus GK, Svindland A, Sjo OH, Hektoen M,
Eknaes M, et al: CpG island methylator phenotype identifies high
risk patients among microsatellite stable BRAF mutated colorectal
cancers. Int J Cancer. 141:967–976. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guastadisegni C, Colafranceschi M, Ottini
L and Dogliotti E: Microsatellite instability as a marker of
prognosis and response to therapy: A meta-analysis of colorectal
cancer survival data. Eur J Cancer. 46:2788–2798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marisa L, de Reyniès A, Duval A, Selves J,
Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D,
Ayadi M, et al: Gene expression classification of colon cancer into
molecular subtypes: Characterization, validation, and prognostic
value. PLoS Med. 10:e10014532013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peng H, Long F and Ding C: Feature
selection based on mutual information: Criteria of max-dependency,
max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach
Intell. 27:1226–1238. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou Y, Zhang N, Li BQ, Huang T, Cai YD
and Kong XY: A method to distinguish between lysine acetylation and
lysine ubiquitination with feature selection and analysis. J Biomol
Struct Dyn. 33:2479–2490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao TH, Jiang M, Huang T, Li BQ, Zhang N,
Li HP and Cai YD: A novel method of predicting protein disordered
regions based on sequence features. Biomed Res Int.
2013:4143272013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niu B, Huang G, Zheng L, Wang X, Chen F,
Zhang Y and Huang T: Prediction of substrate-enzyme-product
interaction based on molecular descriptors and physicochemical
properties. Biomed Res Int. 2013:6742152013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang N, Wang M, Zhang P and Huang T:
Classification of cancers based on copy number variation
landscapes. Biochim Biophys Acta. 1860:2750–2755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu L, Chen L, Zhang YH, Wei L, Cheng S,
Kong X, Zheng M, Huang T and Cai YD: Analysis and prediction of
drug-drug interaction by minimum redundancy maximum relevance and
incremental feature selection. J Biomol Struct Dyn. 35:312–329.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang N, Huang T and Cai YD:
Discriminating between deleterious and neutral non-frameshifting
indels based on protein interaction networks and hybrid properties.
Mol Genet Genomics. 290:343–352. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shu Y, Zhang N, Kong X, Huang T and Cai
YD: Predicting A-to-I RNA editing by feature selection and random
forest. PLoS One. 9:e1106072014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li BQ, You J, Huang T and Cai YD:
Classification of non-small cell lung cancer based on copy number
alterations. PLoS One. 9:e883002014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang PW, Chen L, Huang T, Zhang N, Kong
XY and Cai YD: Classifying ten types of major cancers based on
reverse phase protein array profiles. PLoS One. 10:e01231472015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang T, Shu Y and Cai YD: Genetic
differences among ethnic groups. BMC Genomics. 16:10932015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang T, Wang M and Cai YD: Analysis of
the preferences for splice codes across tissues. Protein Cell.
6:904–907. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen L, Zhang YH, Huang T and Cai YD:
Identifying novel protein phenotype annotations by hybridizing
protein-protein interactions and protein sequence similarities. Mol
Genet Genomics. 291:913–934. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Chen L, Wang S, Zhang Y, Kong X,
Huang T and Cai YD: A computational method using the random walk
with restart algorithm for identifying novel epigenetic factors.
Mol Genet Genomics. 293:293–301. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li L, Wang Y, An L, Kong X and Huang T: A
network-based method using a random walk with restart algorithm and
screening tests to identify novel genes associated with Menière's
disease. PLoS One. 12:e01825922017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen L, Chu C, Kong X, Huang G, Huang T
and Cai YD: A hybrid computational method for the discovery of
novel reproduction-related genes. PLoS One. 10:e01170902015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue):. D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen L, Zhang YH, Li J, Wang S, Zhang Y,
Huang T and Cai YD: Deciphering the relationship between obesity
and various diseases from a network perspective. Genes (Basel).
8(pii): E3922017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen L, Pan H, Zhang YH, Feng K, Kong X,
Huang T and Cai YD: Network-based method for identifying
co-regeneration genes in bone, dentin, nerve and vessel tissues.
Genes (Basel). 8(pii): E2522017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, Yang J, Huang T, Shu Y and Chen
L: Identification of novel proliferative diabetic retinopathy
related genes on protein-protein interaction network.
Neurocomputing. 217:63–72. 2016. View Article : Google Scholar
|
|
34
|
Yang J, Huang T, Song WM, Petralia F,
Mobbs CV, Zhang B, Zhao Y, Schadt EE, Zhu J and Tu Z: Discover the
network mechanisms underlying the connections between aging and
age-related diseases. Sci Rep. 6:325662016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Yang J, Huang T, Kong X, Lu L and
Cai YD: Mining for novel tumor suppressor genes using a shortest
path approach. J Biomol Struct Dyn. 34:664–675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen L, Huang T, Zhang YH, Jiang Y, Zheng
M and Cai YD: Identification of novel candidate drivers connecting
different dysfunctional levels for lung adenocarcinoma using
protein-protein interactions and a shortest path approach. Sci Rep.
6:298492016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Niu B, Lu Y, Lu J, Chen F, Zhao T, Liu Z,
Huang T and Zhang Y: Prediction of enzyme's family based on
protein-protein interaction network. Current Bioinform. 10:16–21.
2015. View Article : Google Scholar
|
|
38
|
Chen L, Chu C, Lu J, Kong X, Huang T and
Cai YD: A computational method for the identification of new
candidate carcinogenic and non-carcinogenic chemicals. Mol Biosyst.
11:2541–2550. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang T, Liu CL, Li LL, Cai MH, Chen WZ,
Xu YF, O'Reilly PF, Cai L and He L: A new method for identifying
causal genes of schizophrenia and anti-tuberculosis drug-induced
hepatotoxicity. Sci Rep. 6:325712016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hofree M, Shen JP, Carter H, Gross A and
Ideker T: Network-based stratification of tumor mutations. Nat
Methods. 10:1108–1115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alhopuro P, Sammalkorpi H, Niittymäki I,
Biström M, Raitila A, Saharinen J, Nousiainen K, Lehtonen HJ,
Heliövaara E, Puhakka J, et al: Candidate driver genes in
microsatellite-unstable colorectal cancer. Int J Cancer.
130:1558–1566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Parsons MT, Buchanan DD, Thompson B, Young
JP and Spurdle AB: Correlation of tumour BRAF mutations and MLH1
methylation with germline mismatch repair (MMR) gene mutation
status: A literature review assessing utility of tumour features
for MMR variant classification. J Med Genet. 49:151–157. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang M, Zhao Y and Zhang B: Efficient test
and visualization of multi-set intersections. Sci Rep. 5:169232015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Forootan M, Tabatabaeefar M, Yahyaei M and
Maghsoodi N: Metabolic syndrome and colorectal cancer: A
cross-sectional survey. Asian Pac J Cancer Prev. 13:4999–5002.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brown DG, Rao S, Weir TL, O'Malia J, Bazan
M, Brown RJ and Ryan EP: Metabolomics and metabolic pathway
networks from human colorectal cancers, adjacent mucosa, and stool.
Cancer Metab. 4:112016. View Article : Google Scholar : PubMed/NCBI
|