Introduction
Retinoblastoma (RB) is a severe ophthalmic disease.
It is the most common type of intraocular malignant tumor among
infants and damages eyesight, ophthalmic tissues and reduces
quality of life. As an embryonic malignant tumor, RB originates
from the primitive nuclear stem cells of the nuclear layer of the
retina. It accounts for 4% of childhood malignant tumors (1). This disease can lead to impaired
vision and blindness (2). At
present, there are mainly three treatments for RB as follows:
Topical, surgical and systemic. Topical treatment, including
cryotherapy, laser photocoagulation, transpupillary thermotherapy,
local radiotherapy, external radiation therapy may be applied to
rescue the eyeball by direct destruction of the tumor. Systemic
chemotherapy is widely applied in the systemic therapy of RB.
Surgical treatment mainly refers to the enucleation of eyeballs and
orbital exenteration surgery (3–8). These
therapies have notable side effects and defects; however, there is
still no effective method and medication to treat RB. Gene therapy
is considered to be a potential cure of the disease (9–12).
Suicide gene therapy against tumor cells results in the expression
of a metabolic enzyme gene through transgenic manipulation; the
expressed metabolic enzyme can convert non-toxic compounds
(prodrug) into cytotoxic drugs that can kill cells, thereby tumor
cells are eliminated selectively (13–15).
The Herpes simplex virus type 1 thymidine kinase
(HSV-TK)/ganciclovir (GCV) system has been employed in extensive
studies, and is also the most widely studied suicide gene. HSV-TK
is a specific product of type 1 HSV (16,17).
HSV-TK can phosphorylate GCV to bisphosphonates-GCV (GCV-DP) and
triphosphate-GCV (GCV-TP); GCV-TP is a competitive inhibitor of DNA
polymerase that can terminate DNA chain elongation and eventually
cause cell death, thereby inducing mammalian cell toxicity
(18). The HSV-TK/GCV system also
leads to irreparable DNA breaks and inhibits the homologous repair
of genes, greatly enhancing cytotoxicity (18,19).
When HSV-TK-transfected and HSV-TK-negative cells were co-cultured
and treated with GCV, HSV-TK-positive cell apoptosis was detected;
however, apoptosis may be induced in HSV-TK-negative cells. The
bystander effect further suggests that the HSV-TK/GCV system may
exhibit notable antitumor effects (20,21).
However, the exact molecular mechanisms underlying the antitumor
effects of the HSV-TK/GCV system have not been elucidated; in
particular, the antitumor mechanism of RB requires more detailed
research and further investigation.
Autophagy is a specific cell response to a range of
stressors, including nutritional deficiency, growth factor
deprivation or other genetic mutations-induced stresses (22–24).
This process constitutes the degradation of endogenous proteins
within lysosomes to remove damaged old organelles, such as damaged
mitochondria, and contributes to the maintenance of cell
self-renewal and homeostasis (22,25).
The 2016 Nobel Prize in Physiology or Medicine was awarded to cell
biologist Yoshinori Ohsumi, who identified and characterized the
autophagy machinery in yeast in 1993 (26). Numerous studies have shown that
autophagy is associated with many diseases, including
neurodegenerative diseases (27),
autoimmunity (28), heart (29) and metabolic diseases (30) and cancer (31). In recent years, autophagy has been
reported to serve an important role in tumor formation,
proliferation, and migration (32–36).
Autophagy inhibitors, such as chloroquine, have demonstrated marked
antitumor effects in breast, colon, and non-small cell lung cancers
(37–40). Therefore, the present study aimed to
investigate whether the HSV-TK/GCV system may serve an antitumor
role in human retinal tumor cells and this role may be mediated by
autophagy. In the present study, we transfected HSV-TK into two
retinal tumor cell lines and then treated with GCV. The results
revealed that HSV-TK/GCV may induce notable retinal tumor cell
cytotoxicity. Additionally, our results also showed that HSV-TK/GCV
did not inhibit autophagy by classic mTOR pathways. It was reported
that the MAPK signaling pathway is closely associated with
autophagy (41). This indicated
that HSV-TK/GCV may induce cell cytotoxicity by affecting autophagy
through the MAPK/ERK signaling pathway. The findings of the present
study also revealed that HSV-TK/GCV may significantly enhance the
levels of phospho-ERK1/2, suggesting that cell cytotoxicity induced
by HSV-TK/GCV may inhibit autophagy via the activation of
MAPK/ERK.
In conclusion, our results indicated that HSV-TK/GCV
could notably induce the cytotoxicity of RB cells, and its
molecular mechanism may affect autophagy by activating MAPK/ERK.
These results may provide novel insights for the advanced treatment
of retinal tumors via an optimized HSV-TK/GCV system.
Materials and methods
Plasmid constructs
To generate pLenO-GTP-HSV-TK, the HSV-TK
oligonucleotides were synthesized according to GenBank (gene ID:
1487307), and then cloned to lentivirus vector pLenO-GTP via
EcoRI and BamHI; mCherry-hLC3B-pcDNA3.1 was a gift
from David Rubinsztein (Addgene plasmid cat. no. 40827) (42), and this plasmid was transfected into
293 cells to observe the effect of HSV-TK/GCV system on autophagic
flux. All plasmids were confirmed by sequencing (The Beijing
Genomics Institute, Beijing, China).
Lentiviral particles preparation
Transient production of the lentiviral vector
particles was conducted in 293 cells by transfecting vector plasmid
pLenO-GTP–HSV-TK, generation packaging plasmids and envelope
glycoprotein pRsv-REV, pMDlg-pRRE and pMD2G. After 72 h,
supernatants containing viral particles were harvested and
centrifuged at 4,000 × g at 4°C for 10 min. The supernatant was
filtered with a 0.45-µm membrane and then centrifuged at 25,000 × g
at 4°C for 2 h. The lentiviral particles were suspended with
ice-cold DMEM and stored at −80°C.
Cell culture, transfection and
drugs
Retinoblastoma cell lines HXO-RB44 and Y79 were
cultured in RPMI-1640 Medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS)
(Gibco; Thermo Fisher Scientific, Inc.) with penicillin (100 U/ml)
and streptomycin (100 g/ml). 293 and HeLa cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) with penicillin (100 U/ml) and
streptomycin (100 g/ml). To obtain expressing HSV-TK HXO-RB44
cells, HXO-RB44 cells were transfected with HSV-TK
(pLenO-GTP-HSV-TK). After 48 h post-transfection, the medium was
replaced with RPMI-1640 containing penicillin (100 U/ml),
streptomycin (100 g/ml) and puromycine (4 µg/ml). 293 and HeLa
cells were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA); Y79 and HXO-RB44 retinoblastoma cell
line were obtained from Shanghai Ninth People's Hospital (Shanghai,
China). The HSV-TK positive cells were treated with GCV at 0, 10,
20 or 40 µg/ml. GCV was purchased from Sigma-Aldrich, ERK inhibitor
U0126 was purchased from Selleck Chemicals (Houston, TX, USA),
Bafilomycine A1 and Torin1 were purchased from Cell Signaling
Technology, (Danvers, MA, USA). To investigate the effect of
HSV-TK/GCV system on autophagic flux, HSV-TK positive HXO-RB44
cells were pretreated with Torin1 (250 nM) or Baf A1 (100 nM) for
12 h at room temperature, subsequently treated with GCV at 20 µg/ml
for 48 h. To investigate effects of ERK inhibitor U0126 on
HSV-TK/GCV system, HSV-TK positive HXO-RB44 cells were pretreated
with U0126 (10 µM) for 4 h at room temperature and subsequently
treated with GCV at 20 µg/ml for 48 h, and then the cell viability
was measured by MTT and compare with the control group that no
treatment with inhibitor.
Immunoblot analysis
he cells were harvested after transfection 48 h,
treated with GCV and were lysed in cell lysis buffer [25 mM
Tris-HCl (pH 7.6), 1% NP-40, 150 mM NaCl and 1%
sodiumdeoxycholate]; a protease inhibitor cocktail was also applied
(Roche Diagnostics, Basel, Switzerland). The proteins were
separated by SDS-PAGE and then transferred onto a polyvinylidene
difluoride membrane (Millipore) for immunoblotting. Immunoblot
analysis was performed with the following primary antibodies:
monoclonal anti-p62 (1:500; cat. no. ab56416), polyclonal anti-LC3
(1:5,000; cat. no. ab51520) was purchased from Abcam (Cambridge,
UK), monoclonal anti-GAPDH (1:20,000; cat. no. MAB374) was
purchased from Millipore, polyclonal anti-phospho-p70S6K antibody
(1:1,000; cat. no. 9234), anti-p70S6K (1:1,000; cat. no. 2708),
anti-phospho-Erk1/2 (1:1,000; cat. no. 4370), anti-Erk1/2 (1:1,000;
cat. no. 4695), anti-phospho-p38 (1:1,000; cat. no. 4511), anti-p38
(1:1,000; cat. no. 8690), anti-phospho-JNK1/2 (1:500; cat. no.
4668), anti-JNK1/2 (1:500; cat. no. 9252), anti-phospho-mTOR
(1:300; cat. no. 5536), anti-mTOR (1:300; cat. no. 2983), anti-AMPK
(1:1,000; cat. no. 5832), anti-phospho-AMPK (1:1,000; cat. no.
2535) antibody were purchased from Cell Signaling Technology,
Inc.
Cell imaging
293 cells were washed with pre-warmed PBS and then
fixed with 4% paraformaldehyde in PBS at room temperature for 5
min, the cells were observed using fluorescence microscopy (Nikon).
HeLa cells transfected with HSV-TK and treated with GCV were
observed 24 or 48 h later using fluorescence microscopy
(Nikon).
Cell death and viability assay
HSV-TK-positive RB cells HXO-RB44 and Y79 were
treated with GCV and analyzed with an MTT assay as follows: The
cells were incubated with 0.5 mg/ml MTT [3-(4,5)-dimethylthiahiazo
(-z-y1)-3,5-di-phenytetrazoliumromide, purchased from
(Sigma-Aldrich; Merck KGaA)] at the concentration of 0.5 mg/ml in
RPMI-1640 medium without phenol red for 3 h at room temperature. As
HXO-RB44 and Y79 were in suspension, the cells were collected via
centrifugation at 1,000 rpm for 5 min; the media was then
discarded. The cells were dissolved in dimethyl sulfoxide and
subsequently centrifuged at 12,000 rpm for 5 min. The optical
density was measured with a photometer at 570 nm, and background at
630 nm was subtracted. The quantitative data were normalized to the
control that HSV-TK-negative cells not treated with GCV and the
ratios are presented as the mean ± standard error of the mean. All
quantitative data were analyzed from three independent
experiments.
Small interfering RNA (siRNA)
transfection
The siRNA specific for Beclin1
(CUAAGGAGCUGCCGUUAUAUU), ATG5 (GGAAUAUCCUGCAGAAGAAUU) or control
siRNA were purchased from Shanghai GenePharma Co., Ltd. (Shanghai,
China) (43). Cells were
transfected with siRNA using RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Cells were then subjected to immunoblot analysis after incubated
for 48 h.
Reverse transcription polymerase chain
reaction (RT-PCR)
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen) and reverse transcribed to complementary DNA
according to the manufacturer's instructions (RR037; Takara).
RT-PCR analysis was performed to identify whether HSV-TK was stably
expressed in cells using a PCR detection system (Applied
Biosystems). The cDNA was amplified by PCR as follows (50 µl)
(R005Q; Takara): cDNA template 50 ng; Pyrobest™ DNA Polymerase 0.2
µl; dNTP mix (0.5 mM each); 10X Pyrobest Buffer II
(Mg2+plus, 10 mM); primer (0.5 µM); denaturation
temperature: 95°C, 30 sec; annealing temperature 56°C, 30 sec;
extension reaction: 72°C, 40 sec; 35 cycles. The products were
separated on a 1% agarose gel and stained with ethidium bromide.
The following primers were used: 5′-ATGACAAGCGCCCAGATA-3′ and
5′-AGGGTAAATAACGTGTCC-3′ for a 512 bp target sequence.
Statistical analysis
Statistical analyses were performed using a
two-tailed Student's t-test for the comparison of two groups. The
comparison of multiple groups were analyzed using one-way analysis
of variance (ANOVA) or two-way ANOVA depending on comparison
variables, followed by a Tukey's pot hoc analysis as indicated
(GraphPad Prism 6; GraphPad Software, Inc., La Jolla, CA, USA).
Data were expressed as the mean ± standard deviation. P<0.05 was
considered statistically significant.
Results
HSV-TK is stably expressed in retinal
tumor cells and other cells
We synthesized HSV-TK sequence fragments and cloned
them into lentiviral vector for transfection into cells. The target
gene was successfully constructed and identified by RT-PCR,
restriction endonuclease-mediated identification and PCR sequencing
(Fig. 1A and B).
Subsequently, plasmids containing HSV-TK were
transfected to HXO-RB44 cells, and the effect of transfection was
observed by fluorescence after 48 h (Fig. 1C). Simultaneously, to identify
stable expression in cells, total RNA was extracted from the
transfected cells, which revealed that the HSV-TK sequence can be
detected via RT-PCR (Fig. 1D).
These findings show that HSV-TK can be stably expressed in HXO-RB44
cells. Similar results were observed in other cells such as Y79,
HeLa and 293 which were transfected with HSV-TK (data not shown).
These results suggest that the HSV-TK constructed in the present
study can be stably expressed in retinal tumor cell Y79 and other
cells, including HeLa and 293, which may be used in future
investigations.
HSV-TK/GCV can significantly induce
retinal tumor cell death as well as other tumor cells
Many studies have reported that HSV-TK/GCV can
induce the apoptosis of a variety of tumor cell types (19,62).
In this study, to investigate whether HSV-TK/GCV could also induce
retinal tumor cell death, HSV-TK were transfected into the tumor
cell line HXO-RB44, which was treated with GCV at 10, 20 or 40
µg/ml for 24 and 48 h respectively; cell viability was then
detected with an MTT assay. The results demonstrated that after
HSV-TK transfection, GCV treatment may significantly reduce cell
viability and as time and GCV concentration increase (Fig. 2A and B). HSV-TK/GCV-inducing cell
death was reported to be dependent on GCV concentration and time.
However, the cell viability of the control group (without HSV-TK
transfection) did not significantly decrease after GCV treatment
(Fig. 2A and B). Similar results
were also obtained in another retinal tumor cell line, Y79
(Fig. 2E). Additionally, HeLa cells
transfected with HSV-TK and treated with GCV, exhibited significant
induction of cell death (Fig. 2C and
D). These results indicate that HSV-TK/GCV may significantly
induce the apoptosis of retinal tumor cells and other types of
tumor cells.
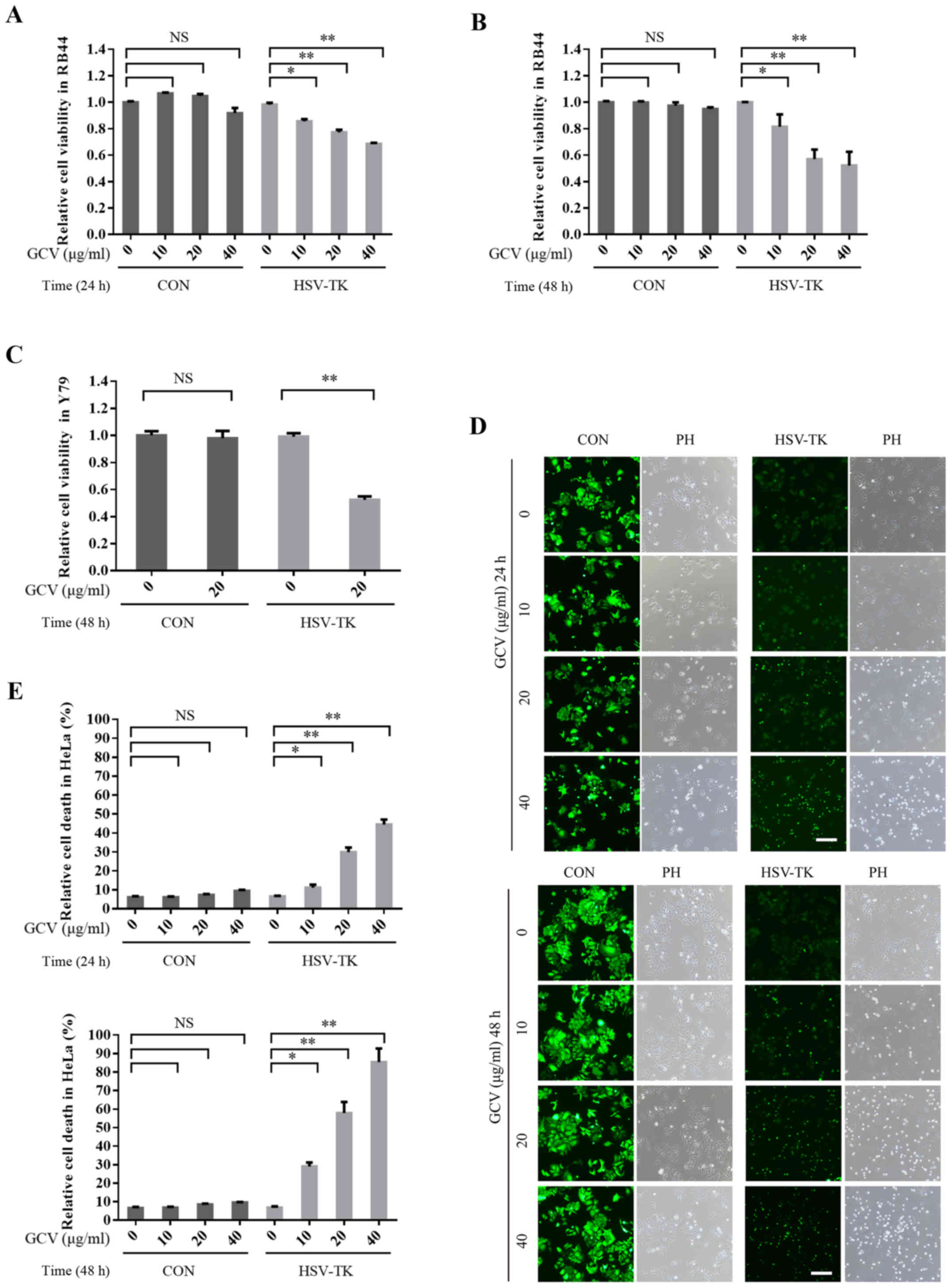 | Figure 2.HSV-TK/GCV can significantly induce
apoptosis of retinal tumor cells, as well as other tumor cells. (A)
HSV-TK-positive (HSV-TK: pLenO-GTP-HSV-TK) or negative (CON:
pLenO-GTP) HXO-RB44 cells were treated with GCV at 0, 10, 20 or 40
µg/ml respectively. After 24 h post-transfection, cell viability
was measured by MTT assay. (B) HSV-TK-positive (HSV-TK:
pLenO-GTP-HSV-TK) or negative (CON: pLenO-GTP) HXO-RB44 cells were
treated with GCV at 0, 10, 20 or 40 µg/ml, respectively for 48 h.
Cell viability was also measured by MTT assay. (C) Y79 cells
transfected with CON or HSV-TK for 48 h; cells were then treated
with GCV at 20 µg/m for 48 h. Cell viability were measured by MTT
assay. (D and E) HeLa cells transfected CON (pLenO-GTP) or HSV-TK
(pLenO-GTP-HSV-TK) for 48 h, then the cells were treated with GCV
at 0, 10, 20 or 40 µg/ml respectively. Cell death was measured
after 24 or 48 h. The positive cells were colored green. The
quantitative data are indicated as the means ± SD. *P<0.05,
**P<0.01. NS, no significance; GCV, ganciclovir; HSV-TK, Herpes
simplex virus type 1 thymidine kinase. |
HSV-TK/GCV can upregulate
autophagy-associated proteins
Autophagy is important for maintaining cell growth
and normal function (30). When
autophagy is abnormal, the autophagic abnormalities may lead to
cell dysfunction, such as protein degradation disorders, cell
growth block or cell cycle changes (23–25).
Therefore, the present study aimed to investigate whether retinal
tumor cell death induced by HSV-TK/GCV may be mediated by
autophagy. HSV-TK-positive HXO-RB44 cells were treated with 0, 10,
20 or 40 µg/ml GCV for 24 or 48 h respectively, the level of LC3
type 2 (LC3II) was significantly upregulated (Fig. 3A, C, D and F). The adaptor protein
p62/SQSTM1 is an autophagy substrate that is selectively degraded
via autophagy and serves an important role in autophagy (44–47).
The results show that p62 associated with autophagy also increased
notably (Fig. 3A, B, D and E). The
same results were observed in Y79 cells (Fig. 3G). To further explore the effects of
HSV-TK/GCV on autophagy, the expression levels of LC3 type 2 were
observed after ATG5 or Beclin1 knockdown in HXO-RB44 cells
transfected with HSV-TK or non-transfected cells, and treated with
or without GCV. Knockdown ATG5 in the HSV-TK positive HXO-RB44
cells, when the cytosolic LC3 type (LC3I) to autophagosomal
membramne-LC3 (LC3II) prevented, the results showed that the cells
treated with GCV resulted in a decrease in the expression levels of
LC3II (Fig. 3H). In addition, the
knockdown of Beclin1 in HXO-RB44 cells expressing HSV-TK revealed
similar results. However, knockdown ATG5 or Beclin1 in HXO-RB44
cells which were HSV-TK negative did not affect LC3II expression
levels regardless of whether GCV was administered or not (Fig. 3H). By manipulating autophagic flux,
the present study reported that HSV-TK/GCV-induced cytotoxicity
maybe mediated by inhibiting autophagy. To further observe
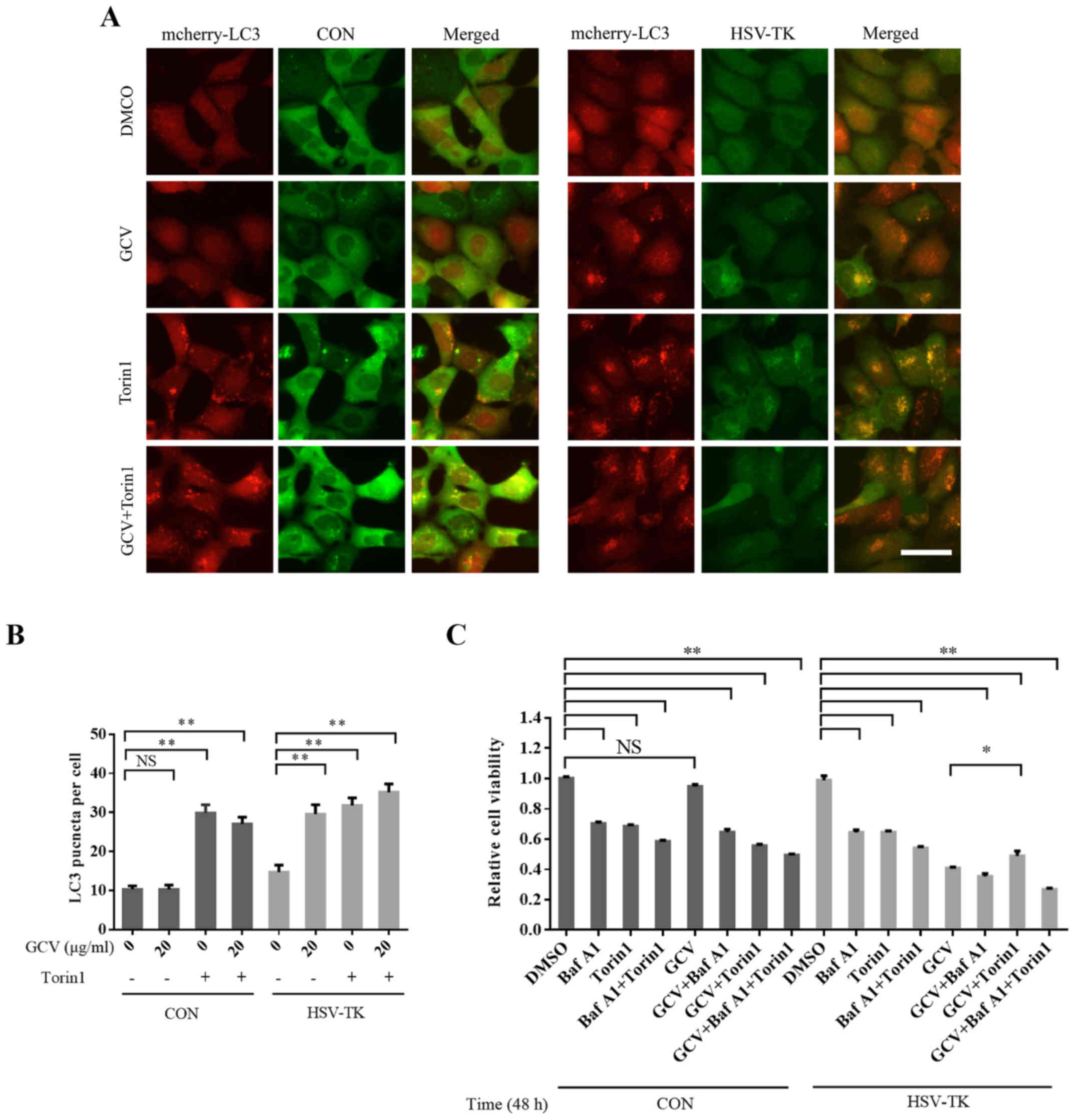
HSV-TK/GCV-associated effects on autophagy, HSV-TK and mcherry-LC3
were co-transfected into 293 cells, which were then treated with
GCV for 48 h at 10 µg/ml. It was observed that LC3 aggregation
increased significantly (Fig. 4A and
B), indicating that autophagy was affected. Torin1 activates
autophagy via the inhibition of mTOR. While pretreated
HSV-TK-positive HXO-RB44 cells with Torin1 (250 nM) for 12 h, and
then removed Torin1, subsequently treated with GCV, the results
suggested that Torin1 may partly attenuate the reduced cell
viability induced by HSV-TK/GCV (Fig.
4C). These results suggest that HSV-TK/GCV-inducing retinal
tumor cell death may be mediated by inhibition of autophagy.
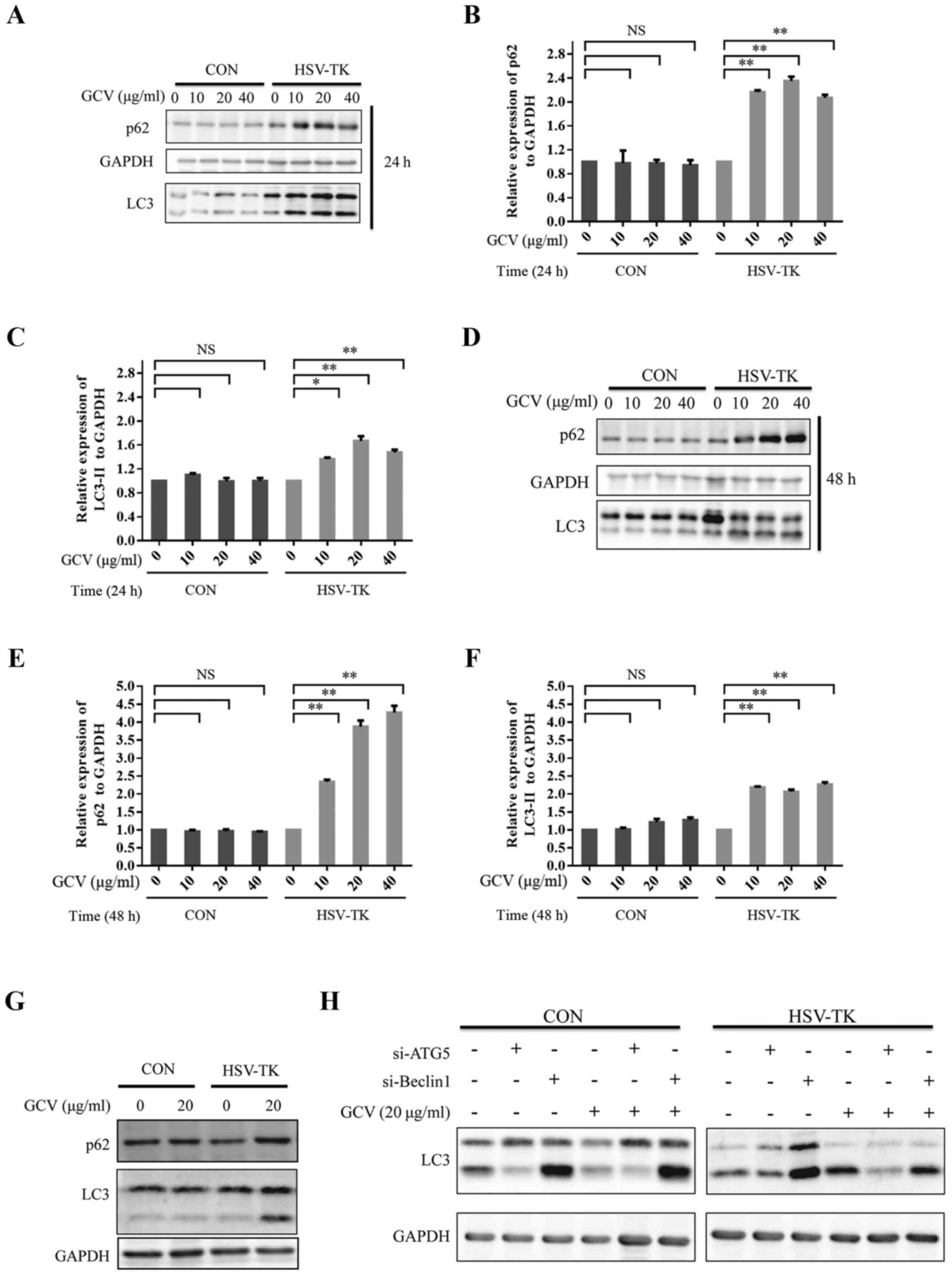 | Figure 3.HSV-TK/GCV can upregulate
autophagy-associated proteins. (A) CON (pLenO-GTP) or HSV-TK
(pLenO-GTP-HSV-TK) were transfected to HXO-RB44 cells. After 48 h
later the cells were incubated with GCV at 0, 10, 20 or 40 µg/ml,
respectively for 24 h. The cell lysates were then collected and
subjected to immunoblotting. (B and C) p62 and LC3II expression
levels in (A) were quantified. (D) HXO-RB44 cells transfected in
the same manner as described in (A), and then treated with GCV at
0, 10, 20 or 40 µg/ml, respectively for 48 h. The cell lysates were
then collected and subjected to immunoblotting. (E and F) p62 and
LC3II expression levels in (D) were quantified. (G) CON or HSV-TK
were transfected to Y79 cells. After 48 h later the cells were
incubated with GCV at 20 µg/ml for 48 h. The cell lysates were then
collected and subjected to immunoblotting. (H) The protein levels
from three independent experiments were quantified. The
quantitative data are indicated as the means ± SD. NS, no
significance, *P<0.05, **P<0.01. GCV, ganciclovir; HSV-TK,
Herpes simplex virus type 1 thymidine kinase. |
HSV-TK/GCV induces cell cytotoxicity
by specifically activating MAPK/ERK to inhibit autophagy
To further explore the molecular mechanism of
HSV-TK/GCV autophagy activation, autophagy-associated signaling
pathways were investigated. mTOR serves a central role in the
regulation of autophagy (48). When
HSV-TK-positive HXO-RB44 cells were treated with 20 µg/ml GCV for
48 h, the results indicated that compared with in nontransfected
cells, GCV treatment or transfection with HSV-TK but without GCV
treatment exhibited no significant changes in phospho-mTOR and
P70S6K substrate levels, suggesting that HSV-TK/GCV may not affect
autophagy through the mTOR signaling pathway (Fig. 5A).
MAPK plays an important role in maintaining cell
functions, such as cell proliferation (49), growth and differentiation (50,51),
and other functions (52). Many
studies have also reported that MAPK is closely related to
autophagy (41,53,54).
The results of the present showed that the levels of phosphorylated
ERK were significantly upregulated in HXO-RB44 cells transfected
with HSV-TK and treated with GCV, compared with in the groups
without transfection treated with GCV or with transfection but
without GCV treatment. Other MAPK-associated signaling pathways,
including p38 and JNK, did not significantly change (Fig. 5B). Similar results were observed in
the Y79 cells (data not shown). To further verify the findings of
the present study, the MAPK/ERK signaling pathway was inhibited
with U0126, a specific MEK inhibitor; whether cell cytotoxicity
could be attenuated was investigated. The present study
demonstrated that U0126 may partly but not completely attenuate
reductions in cell viability (Fig.
5C). These results indicate that HSV-TK/GCV may induce cell
death by inhibiting autophagy via specific MAPK/ERK activation
(Fig. 5D).
Discussion
Numerous articles have previously reported that
HSV-TK/GCV can significantly induce cell death in a variety of
tumor cells; one of the underlying mechanisms for this cell death
may induce apoptosis through inducing DNA damage (18,21,37).
However, the underlying molecular mechanisms have not been fully
elucidated.
Autophagy is an important process within cells to
maintain normal function. Cells can achieve energy reuse and
self-renewal via the degradation of proteins and damaged organelles
by autophagy (22,23,25,30).
Since the discovery of autophagy, researchers have found that its
dysfunction is closely associated with numerous diseases, including
cancer (27–29,38).
The HSV-TK/GCV system may also induce retinal tumor cell death; the
present study reported that HSV-TK/GCV can induce significant death
of two retinal tumor cell lines, HXO-RB44 and Y79, but also may
induce other cell death, such as HeLa. The present study proposed
that cell death may be mediated by affecting autophagy, and that
LC3II and P62 are key components of autophagy. Our results
suggested that LC3II and P62 expression levels of HXO-RB44 and Y79
cells were upregulated after transfection with HSV-TK and GCV
treatment in a time- and dose-dependent manner. Manipulation of
autophagic flux by knockdown ATG5 or Beclin1 revealed that LC3II of
HSV-TK-positive HXO-RB44 cells treated with GCV decreased compared
with the transfected group without GCV treatment. However,
knockdown ATG5 or Beclin1 in HSV-TK negative HXO-RB44 cells
indicated that LC3-II expression levels did not change in the
presence or absence of GCV treatment. These results suggest that
this cell death may mediated by the inhibition of autophagy. mTOR
that plays a central role in the regulation of autophagy may
inhibit autophagy by the phosphorylation of ATG13 (30,55).
Simultaneously, AMPK can also activate autophagy (56). Our results demonstrated that
HSV-TK/GCV did not inhibit autophagy via the mTOR signaling pathway
and did not affect AMPK activity (Fig.
5A).
MAPK is a mitogen-activated protein kinase, which
can regulate cell growth and differentiation, as well as stress and
inflammatory responses to the external environment, and other
important cellular physiological/pathological processes (28,50–52).
It has been reported that MAPK also regulates autophagy signaling
pathways (41,54). The findings of the present study
show that HSV-TK-positive retinal tumor cells were treated with GCV
exhibited unaffected mTOR activity; the levels of their downstream
substrate P70S6K did not change significantly (Fig. 5A). These results suggest that the
HSV-TK/GCV system may not affect autophagy via the classical mTOR
signaling pathway. In addition, the present study investigated
whether AMPK, MAPK and other signal pathways were affected. The
results demonstrated that the activities of AMPK, MAPK/P38 and
MAPK/JNK were not significantly affected; however, phospho-ERK1/2
expression levels were upregulated (Fig. 5A and B). These results indicated
that HSV-TK/GCV may reduce MAPK/ERK activity to activate autophagy
and induce cell cytotoxicity.
Numerous studies have reported that the HSV-TK1/GCV
system may block DNA chain elongation, leading to cell death
(11,18), and that the toxicity induced by
HSV-TK/GCV may occur via apoptosis (57,58).
In the present study, the molecular mechanism underlying cell
cytotoxicity to increase MAPK/ERK activity for the inhibition of
autophagy was revealed. Additionally, activate autophagy through
Torin1, an inhibitor of mTOR may partially, but not completely
rescue cell cytotoxicity; however, in response to treatment with
autophagy inhibitor Bafilomycine A1 and mTOR inhibitor Torin1, cell
viability was significantly reduced compared with Torin1 or Baf A1
treatment alone (Fig. 4C). In
addition, the effects of MAPK/ERK inhibitor may partly rescue cell
cytotoxicity induced by the HSV-TL/GCV system (Fig. 5C), and may provide further support
of the aforementioned results.
Cell cytotoxicity cannot be rescued by activating
autophagy via treatment with Torin1, because HSV-TK/GCV may
maximally inhibit autophagy, and cannot be reversed by Torin1,
which itself has a certain cytotoxicity. The second reason is that
autophagy is but one of the several ways that HSV-TK/GCV can induce
cell cytotoxicity; however, the extent of the effects of autophagy
on apoptosis is unclear. There are many reports that autophagy is
closely related to cell apoptosis, and they even may be induced by
common upstream signals (59). The
inhibition of autophagy may induce cell apoptosis (59). Therefore, future research may
investigate the association between autophagy and apoptosis in cell
death induced by HSV-TK/GCV. At present, HSV-TK/GCV has been
studied in numerous animals and in clinic (60–62).
The rAAV-HSV-TK system has demonstrated inhibition of tumor cell
growth with strong antitumor efficacy in mice models, and may be
considered as a potential strategy for the treatment of bladder
carcinoma (63). Similarly, as
overserved in a mouse xenograft model of lung cancer, HSV-TK/GCV
therapy may reduce tumor size (64). At the same time, HSV-TK was also
demonstrated to be a promising mode of therapy in combination with
other treatments (65). Few studies
have reported that HSV-TK/GCV may induce retinal tumor cell
toxicity; however, investigation has not been conducted within
relevant animal models. The majority of the experiments in the
present study were performed in vitro; further study is
required to explore the molecular mechanism in vivo and
clinical research of the association between HSV-TK/GCV for
retinoblastoma may be conducted. In conclusion, the results
suggested that further investigation into other mechanisms
underlying HSV-TK/GCV-medicated cell cytotoxicity is required to
develop treatments with increased therapeutic effectiveness. The
findings of the present study regarding the effects of HSV-TK/GCV
on autophagy may improve current understanding on its therapeutic
mechanisms and contribute to research developments in clinical and
non-clinical studies.
Acknowledgements
We thank Dr David Rubinsztein for providing us with
the plasmids mCherry-hLC3B-pcDNA3.1.
Funding
The present study is supported by the Natural
Science Foundation of Ningbo City (no. 2016A610014), and by the
Projects of Medical and Health Technology Development program in
Zhejiang province (no. 2017KY619).
Availability of data and materials
All data generated or analyzed in this study are
included in this published article.
Authors' contributions
QYY, ZSB and BC carried out the research and data
acquisition. ZSB drafted the manuscript and BC carried out the data
analysis and statistical analysis. QYY designed the research,
manuscript editing and manuscript review. NC, LSC, TY and HM also
provided assistance for the acquisition and detection of cell
samples. All authors read and approved the manuscript and agree to
be accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HSV-TK
|
herpes simplex virus type 1 thymidine
kinase
|
|
GCV
|
ganciclovir
|
|
MAPK
|
mitogen-activated protein kinase
|
|
Baf A1
|
bafilomycine A1
|
|
GCV-DP
|
bisphosphonates-GCV
|
|
GCV-TP
|
triphosphate-GCV
|
References
|
1
|
Abramson DH: Retinoblastoma in the 20th
century: Past success and future challenges the Weisenfeld lecture.
Invest Ophthalmol Vis Sci. 46:2683–2691. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodriguezgalindo C, Wilson MW, Chantada G,
Fu L, Qaddoumi I, Antoneli C, Leal-Leal C, Sharma T, Barnoya M,
Epelman S, et al: Retinoblastoma: One world, one vision.
Pediatrics. 122:e763–e770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bornfeld N, Schüler A, Bechrakis N, Henze
G and Havers W: Preliminary results of primary chemotherapy in
retinoblastoma. Klin Padiatr. 209:216–221. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shields CL and Shields JA: Recent
developments in the management of retinoblastoma. J Pediatr
Ophthalmol Strabismus. 36:8–18; quiz 35–36. 1999.PubMed/NCBI
|
|
5
|
Shields CL, Mashayekhi A, Cater J, Shelil
A, Meadows AT and Shields JA: Chemoreduction for retinoblastoma:
Analysis of tumor control and risks for recurrence in 457 tumors.
Trans Am Ophthalmol Soc. 102:35–44. 2004.PubMed/NCBI
|
|
6
|
Abramson DH, Dunkel IJ, Brodie SE, Kim JW
and Gobin YP: A phase I/II study of direct intraarterial
(ophthalmic artery) chemotherapy with melphalan for intraocular
retinoblastoma initial results. Ophthalmology. 115:1398–1404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shields CL and Shields JA: Retinoblastoma
management: advances in enucleation, intravenous chemoreduction,
and intra-arterial chemotherapy. Curr Opin Ophthalmol. 21:203–212.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodriguez-Galindo C, Chantada GL, Haik BG
and Wilson MW: Treatment of retinoblastoma: Current status and
future perspectives. Curr Treat Options Neurol. 9:294–307. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu HJ, Zhou Y, Ji W, Perng GS, Kruzelock
R, Kong CT, Bast RC, Mills GB, Li J and Hu SX: Reexpression of the
retinoblastoma protein in tumor cells induces senescence and
telomerase inhibition. Oncogene. 15:2589–2596. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayashi N, Ido E, Ohtsuki Y and Ueno H: An
experimental application of gene therapy for human retinoblastoma.
Invest Ophthalmol Vis Sci. 40:265–272. 1999.PubMed/NCBI
|
|
11
|
Cullinan AE, Lindstrom MJ, Sabet S, Albert
DM and Brandt CR: Evaluation of the antitumor effects of Herpes
simplex virus lacking ribonucleotide reductase in a murine
retinoblastoma model. Current Eye Res. 29:167–172. 2004. View Article : Google Scholar
|
|
12
|
Marshall E: Gene therapy a suspect in
leukemia-like disease. Science. 298:34–35. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hossain JA, Ystaas LR, Mrdalj J, Välk K,
Riecken K, Fehse B, Bjerkvig R, Grønli J and Miletic H: Lentiviral
HSV-Tk.007 mediated suicide gene therapy is not toxic for normal
brain cells. J Gene Med. 18:234–243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Howard BD, Boenicke L, Schniewind B,
Henne-Bruns D and Kalthoff H: Transduction of human pancreatic
tumor cells with vesicular stomatitis virus G-pseudotyped
retroviral vectors containing a herpes simplex virus thymidine
kinase mutant gene enhances bystander effects and sensitivity to
ganciclovir. Cancer Gene Ther. 7:927–938. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kenney S and Pagano JS: Viruses as
oncolytic agents: a new age for ‘therapeutic’ viruses? J Natl
Cancer Inst. 86:1185–1186. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lou E: Oncolytic herpes viruses as a
potential mechanism for cancer therapy. Acta Oncol. 42:660–671.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moolten FL: Tumor chemosensitivity
conferred by inserted herpes thymidine kinase genes: Paradigm for a
prospective cancer control strategy. Cancer Res. 46:5276–5281.
1986.PubMed/NCBI
|
|
18
|
Thust R: Ganciclovir-induced apoptosis in
HSV-1 thymidine kinase expressing cells: critical role of DNA
breaks, Bcl-2 decline and caspase-9 activation. Oncogene.
21:2141–2153. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beltinger C, Fulda S, Kammertoens T, Meyer
E, Uckert W and Debatin KM: Herpes simplex virus thymidine
kinase/ganciclovir-induced apoptosis involves ligand-independent
death receptor aggregation and activation of caspases. P Natl Acad
Sci USA. 96:8699–8704. 1999. View Article : Google Scholar
|
|
20
|
Wygoda MR, Wilson MR, Davis MA, Trosko JE,
Rehemtulla A and Lawrence TS: Protection of herpes simplex virus
thymidine kinase-transduced cells from ganciclovir-mediated
cytotoxicity by bystander cells: The good samaritan effect. Cancer
Res. 57:1699–1703. 1997.PubMed/NCBI
|
|
21
|
Touraine RL, Ishiimorita H, Ramsey WJ and
Blaese RM: The bystander effect in the HSVtk/ganciclovir system and
its relationship to gap junctional communication. Gene Ther.
5:1705–1711. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seino J, Wang L, Harada Y, Huang C, Ishii
K, Mizushima N and Suzuki T: Basal autophagy is required for the
efficient catabolism of sialyloligosaccharides. J Biol Chem.
288:26898–27907. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsukada M and Ohsumi Y: Isolation and
characterization of autophagy-defective mutants of Saccharomyces
cerevisiae. FEBS Lett. 333:169–174. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Menzies FM, Fleming A and Rubinsztein DC:
Compromised autophagy and neurodegenerative diseases. Nat Rev
Neurosci. 16:345–357. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhong Z, Sanchezlopez E and Karin M:
Autophagy, inflammation and immunity: A troika governing cancer and
its treatment. Cell. 166:288–298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shirakabe A, Ikeda Y, Sciarretta S,
Zablocki DK and Sadoshima J: Aging and autophagy in the heart. Circ
Res. 118:1563–1576. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim KH and Lee MS: Autophagy[mdash]a key
player in cellular and body metabolism. Nat Rev Endocrinol.
10:322–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brech A, Ahlquist T, Lothe RA and Stenmark
H: Autophagy in tumour suppression and promotion. Mol Oncol.
3:366–375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
33
|
Macintosh RL, Timpson P, Thorburn J,
Anderson KI, Thorburn A and Ryan KM: Inhibition of autophagy
impairs tumor cell invasion in an organotypic model. Cell Cycle.
11:2022–2029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gavilán E, Sánchez-Aguayo I, Daza P and
Ruano D: GSK-3β signaling determines autophagy activation in the
breast tumor cell line MCF7 and inclusion formation in the
non-tumor cell line MCF10A in response to proteasome inhibition.
Cell Death Dis. 4:e5722013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun K, Guo XL, Zhao QD, Jing YY, Kou XR,
Xie XQ, Zhou Y, Cai N, Gao L, Zhao X, et al: Paradoxical role of
autophagy in the dysplastic and tumor-forming stages of
hepatocarcinoma development in rats. Cell Death Dis. 4:e5012013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Colecchia D, Rossi M, Sasdelli F, Sanzone
S, Strambi A and Chiariello M: MAPK15 mediates BCR-ABL1-induced
autophagy and regulates oncogene-dependent cell proliferation and
tumor formation. Autophagy. 11:1790–1802. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang PD, Zhao YL, Shi W, Deng XQ, Xie G,
Mao YQ, Li ZG, Zheng YZ, Yang SY and Wei YQ: Cell growth
inhibition, G2/M cell cycle arrest, and apoptosis induced by
chloroquine in human breast cancer cell line Bcap-37. Cell Physiol
Biochem. 22:431–440. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sasaki K, Tsuno NH, Sunami E, Tsurita G,
Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. Bmc Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zou Y, Ling YH, Sironi J, Schwartz EL,
Perezsoler R and Piperdi B: The autophagy inhibitor chloroquine
overcomes the innate resistance of wild-type EGFR non-small-cell
lung cancer cells to erlotinib. J Thorac Oncol. 8:693–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kao C, Chao A, Tsai CL, Chuang WC, Huang
WP, Chen GC, Lin CY, Wang TH, Wang HS and Lai CH: Bortezomib
enhances cancer cell death by blocking the autophagic flux through
stimulating ERK phosphorylation. Cell Death Dis. 5:e15102014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jahreiss L, Menzies FM and Rubinsztein DC:
The itinerary of autophagosomes: From peripheral formation to
kiss-and-run fusion with lysosomes. Traffic. 9:574–587. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fung C, Lock R, Gao S, Salas E and Debnath
J: Induction of autophagy during extracellular matrix detachment
promotes cell survival. Mol Biol Cell. 19:797–806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moscat J and Diazmeco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zheng YT, Shahnazari S, Brech A, Lamark T,
Johansen T and Brumell JH: The adaptor protein p62/SQSTM1 targets
invading bacteria to the autophagy pathway. J Immunol.
183:5909–5916. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of p62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010.PubMed/NCBI
|
|
48
|
Sancak Y, Peterson TR, Shaul YD, Lindquist
RA, Thoreen CC, Bar-Peled L and Sabatini DM: The rag GTPases bind
raptor and mediate amino acid signaling to mTORC1. Science.
320:1496–1501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ling MT, Wang X, Ouyang XS, Lee TK, Fan
TY, Xu K, Tsao SW and Wong YC: Activation of MAPK signaling pathway
is essential for Id-1 induced serum independent prostate cancer
cell growth. Oncogene. 21:8498–8505. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cui G, Qin X, Zhang Y, Gong Z, Ge B and
Zang YQ: Berberine differentially modulates the activities of ERK,
p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation
in type 1 diabetic mice. J Biol Chem. 284:28420–28429. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hu Q, Li B, Xu R, Chen D, Mu C, Fei E and
Wang G: The protease Omi cleaves the mitogen-activated protein
kinase kinase MEK1 to inhibit microglial activation. Sci Signal.
5:ra612012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Corcelle E, Djerbi N, Mari M, Nebout M,
Fiorini C, Fénichel P, Hofman P, Poujeol P and Mograbi B: Control
of the autophagy maturation step by the MAPK ERK and p38: Lessons
from environmental carcinogens. Autophagy. 3:57–59. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:242014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wei SJ, Chao Y, Hung YM, Lin WC, Yang DM,
Shih YL, Ch'ang LY, Whang-Peng J and Yang WK: S- and G2-phase cell
cycle arrests and apoptosis induced by ganciclovir in murine
melanoma cells transduced with herpes simplex virus thymidine
kinase. Exp Cell Res. 241:66–75. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wei SJ, Chao Y, Shih YL, Yang DM, Hung YM
and Yang WK: Involvement of Fas (CD95/APO-1) and Fas ligand in
apoptosis induced by ganciclovir treatment of tumor cells
transduced with herpes simplex virus thymidine kinase. Gene Ther.
6:420–431. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gerolami R, Cardoso J, Lewin M, Bralet MP,
Sa Cunha A, Clément O, Bréchot C and Tran PL: Evaluation of HSV-tk
gene therapy in a rat model of chemically induced hepatocellular
carcinoma by intratumoral and intrahepatic artery routes. Cancer
Res. 60:993–1001. 2000.PubMed/NCBI
|
|
61
|
Rainov NG: A phase III clinical evaluation
of herpes simplex virus type 1 thymidine kinase and ganciclovir
gene therapy as an adjuvant to surgical resection and radiation in
adults with previously untreated glioblastoma multiforme. Hum Gene
Ther. 11:2389–2401. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Song JS and Kim HP: Adenovirus-mediated
HSV-TK gene therapy using the human telomerase promoter induced
apoptosis of small cell lung cancer cell line. Oncol Rep.
12:443–447. 2004.PubMed/NCBI
|
|
63
|
Pan JG, Zhou X, Luo R and Han RF: The
adeno-associated virus-mediated HSV-TK/GCV suicide system: A
potential strategy for the treatment of bladder carcinoma. Med
Oncol. 29:1938–1947. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Leinonen HM, Ruotsalainen AK, Määttä AM,
Laitinen HM, Kuosmanen SM, Kansanen E, Pikkarainen JT, Lappalainen
JP, Samaranayake H, Lesch HP, et al: Oxidative stress-regulated
lentiviral TK/GCV gene therapy for lung cancer treatment. Cancer
Res. 72:6227–6235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Colombo F, Barzon L, Franchin E, Pacenti
M, Pinna V, Danieli D, Zanusso M and Palù G: Combined HSV-TK/IL-2
gene therapy in patients with recurrent glioblastoma multiforme:
Biological and clinical results. Cancer Gene Ther. 12:835–848.
2005. View Article : Google Scholar : PubMed/NCBI
|