Introduction
Liver cancer, the most common primary liver tumor,
is the third leading cause of cancer mortality globally (1). Surgical resection, thermal ablation and
liver transplantation are the current treatments for early-stage
liver cancer (2). However, liver
cancer is often diagnosed at an advanced stage with poor prognosis.
Sorafenib, a multikinase inhibitor of mitogen-activated protein
kinase/ERK, vascular endothelial growth factor (VEGF) receptor and
platelet-derived growth factor, is the only approved drug available
for the treatment of advance liver cancer (3). Sorafenib has a good tolerability profile
and treatment leads to a longer median survival time (3). Unfortunately, its activity is limited by
primary and acquired drug resistance (4). Thus far, the mechanisms of sorafenib
resistance are not well established; therefore, it is crucial to
determine the mechanisms associated with resistance and to develop
additional therapeutic agents to enhance the effects of the
drug.
Forkhead box M1 (FoxM1) is a member of the forkhead
box transcription factor family. Previous studies have revealed
that FoxM1 has an important role in the pathogenesis and
progression of human cancers, including liver cancer (5,6).
Suppression of FoxM1 expression can lead to chromosome
disaggregation, mitotic spindle defects, mitotic catastrophe and
cell cycle arrest (7). As a key
regulator of the G1/S and G2/M transition, FoxM1 regulates the
expression of several cell cycle-associated factors, including
p21Cip1, cyclin B, p27Kip1, Aurora B kinase,
survivin, CDC25B and polo-like kinase-1 (8,9).
Additionally, the phosphorylation of FoxM1 promotes its nuclear
translocation and enhances its transcriptional activity during G2/M
(10). Previous research has revealed
that FoxM1 is a credible target for arresting cancer growth and
progression. In tumor-bearing mice, knockdown of FoxM1 can
significantly reduce tumor growth (11). By contrast, the overexpression of
FoxM1 can markedly increase the size of tumors (11). FoxM1 can also promote tumor
angiogenesis by enhancing VEGF expression at the transcriptional
level (12) and suppression of FoxM1
inhibited angiogenesis in liver cancer (13). Additionally, senescence was induced in
FoxM1-knockout embryonic fibroblasts, while
H2O2-induced senescence was reversed by FoxM1
overexpression (14). Recent studies
have indicated that the overexpression of FoxM1 may accelerate the
development of acquired drug resistance (15–17).
However, whether the overexpression of FoxM1 contributes to
sorafenib resistance in liver cancer requires further
investigation.
In the present study, FoxM1 was significantly
upregulated in sorafenib-resistant liver cancer cells. Knockdown of
FoxM1 increased the sorafenib sensitivity of drug-tolerant cells.
Furthermore, a signaling axis, the AKT/activator protein-1
(AP1)/FoxM1 signaling pathway, was revealed to contribute to
sorafenib resistance in liver cancer cells. Suppression of this
axis using AKT inhibitor BEZ-235 increased the sensitivity of
drug-tolerant cells to sorafenib in vitro and in
vivo. The data indicated that the AKT/AP1/FoxM1 signaling axis
is a critical determinant of sorafenib tolerance.
Materials and methods
Cell culture
Human liver cancer cell lines HepG2 and Huh7 cells
were purchased from the American Type Culture Collection (ATCC),
and cultured in high-glucose DMEM with 10% FBS, in a humid
incubator with 5% CO2. The two lines were originally
tested by ATCC and passaged <6 months in the laboratory of
Biochemistry and Molecular Biology of Hainan Medical College.
Establishment of sorafenib-resistant
cells
The IC50 value of liver cancer cells to
sorafenib was initially determined by the 4PL method as previously
described (18). The equation is
expressed as follows: Y=a-d1+(X/c)b+d; where Y is the response, and
X is the concentration. The variable ‘a’ is the bottom of the
curve, and ‘d’ is the top of the curve. The variable ‘b’ is the
slope factor, and ‘c’ is the concentration corresponding to the
response midway between ‘a’ and ‘d’. The cells were cultured in
6-well plates at 1×104 cells/well and incubated with
sorafenib at a concentration just below their respective
IC50 values. The concentration of sorafenib was slowly
increased by 0.25 µmol/l per week. After 6–7 months, two
sorafenib-resistant cell lines, termed ‘HepG2-SR’ and ‘Huh7-SR’,
were obtained and were continuously maintained by culture in the
presence of sorafenib.
Cell viability assay
Cell viability was assayed using a Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.). Briefly, the
cells were seeded in triplicate in 96-well plates and underwent
different treatments (including sorafenib at 5, 10, 15 and 20
µmol/l dose, sorafenib combined FoxM1 siRNA, sorafenib combined
c-jun siRNA and sorafenib combined BEZ-235) for 48 h, and then the
optical density value at 450 nm was detected according to the
manufacturer's instructions.
Western blot analysis
Cells were harvested and whole-cell lysates were
prepared. The protein concentrations were measured using a
bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of
Biotechnology). Subsequently, western blot analysis was performed
as previously described (19).
Anti-FoxM1 (dilution 1:1,000; cat. no. ab180710) and c-jun
(dilution 1:1,000; cat. no. ab31419) primary antibodies were
purchased from Abcam. Phosphorylated (p)-JNK (dilution 1:1,000;
cat. no. 9255), p-AKT (dilution 1:1,000; cat. no. 4060), p-ERK
(dilution 1:1,000; cat. no. 9101) antibodies were obtained from
Cell Signaling Technology, Inc. GAPDH (dilution 1:5,000; cat. no.
sc-137179) antibody was obtained from Santa Cruz Biotechnology,
Inc.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were lysed and total RNA was extracted with
TRIzol reagent (Thermo Fisher) as previously described (19), and the first-strand cDNA was
synthesized using M-MLV transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.). qPCR was performed to detect the level of mRNA
using SYBR qPCR master mix (Takara Biotechnology Co., Ltd.) in a
20-µl volume according to the manufacturer's instructions. FOXM1
was amplified using the forward primer, 5′-GCCATCAACAGCACTGAGAG-3′
and reverse primer, 5′-TGGGGTGAATGGTCCAGAAG-3′. JUN was amplified
using the forward primer, 5′-GAGCTGGAGCGCCTGATAAT-3′ and reverse
primer. 5′-CCCTCCTGCTCATCTGTCAC-3′.
Plasmid construction
To construct reporter and mutant reporter plasmids,
FoxM1 genomic DNA was extracted from HepG2 cells. The promoter
region of human FoxM1 (−2,000 to +100) containing the AP1 binding
site was amplified by PCR using the PrimeSTAR HS DNA polymerase
(Takara Biotechnology Co., Ltd.), and then inserted into the
pGL3-Basic vector (Promega Corp.). The recombinant plasmid was
termed pGL3-FoxM1. The mutant plasmid containing the promoter
without the AP1 binding site was constructed using an overlap
primer and termed pGL3-FoxM1-mu.
Transfection assay
The small interfering RNAs against FoxM1, c-Jun and
control siRNA were obtained from Santa Cruz Biotechnology, Inc. The
cells were separately transfected with FoxM1 or c-jun siRNAs and
control siRNA (Shanghai GenePharma Co., Ltd.) at 50 nM using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Luciferase reporter assay
HepG2 cells were seeded in 48-well plates and
cultured to 70–80% confluence. Then, the cells were co-transfected
with pGL3-FoxM1/or pGL3-FoxM1-mu and monitor plasmid RL-PTK. After
36 h, the cells were lysed, and the Firefly and Renilla
luciferase activities were measured using the Dual-Luciferase
Reporter System (Promega Corp.) according to the manufacturer's
instructions. The transfection experiments were performed at least
three times in triplicate. Data are presented as the fold induction
after normalizing the luciferase activity of the tested sample to
that of the corresponding control sample.
Nuclear protein extraction
Kits for nuclear protein extraction and BCA protein
assay were purchased from Beyotime Institute of Biotechnology.
HepG2 cells were seeded to 6-well plates at a density of
2×106 cells and cultured to 80% confluence followed by
the appropriate treatments. Cells were washed in ice-cold PBS, and
resuspended by pipetting up and down in 200 µl ice-cold cell lysis
buffer [10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 1 mM
dithiothreitol (DTT), 0.4% IGEPAL, 2 µg/ml aprotinin, 1 mM PMSF,
250 µg/ml benzamidine, 2 µg/ml leupeptin]. After incubation on ice
for 15 min, cell lysates were centrifuged at 12,000 × g for 5 min
at 4°C and the supernatants (cytoplasm + membrane extract) were
discarded. Nuclear pellets were then resuspended in 50 µl nuclear
extraction buffer (0.4 M NaCl, 20 mM HEPES pH 7.9, 1 mM EDTA, 1 mM
DTT and 1 mM PMSF). After vigorous shaking at 4°C for 30 min, the
nuclear extracts were centrifuged at 12,000 × g for 10 min at 4°C,
and the supernatants for the nuclear protein were collected. The
protein concentration was quantified using a BCA protein assay
kit.
Electrophoretic mobility shift assay
(EMSA)
The EMSA kit was obtained from Pierce (Thermo Fisher
Scientific, Inc.). Cells were harvested for nuclear extraction
using a nuclear and cytoplasmic extraction kit (Thermo Fisher
Scientific, Inc.). The probe sequences were as follows: AP1:
5′-GACTGGTTGACTAAGTCAAT-3′. Mut AP1: 5′-GACTGGCCTACCGGGTCAAT-3′.
Biotin-5′-end-labeled and unlabeled, sense and antisense
oligonucleotides were synthesized. The oligonucleotides were
annealed to generate double-stranded probes. EMSA was performed by
preincubating 3 µg nuclear extract with a mixture containing 1 µg
poly dI:dC and 2 µl binding buffer on ice for 10 min. Then, 20 fmol
biotin-labeled double-stranded probe was added to the mixture and
incubated at room temperature for another 30 min. Competitive EMSA
experiments were conducted by incorporating excess concentrations
of the unlabeled probe in the pre-incubation step of the assay.
DNA-protein complexes were resolved on a non-denaturing 5%
polyacrylamide gel in 0.5X Tris-borate-EDTA. After electrophoresis,
the samples were transferred onto Biodyne B precut modified nylon
membranes (Thermo Fisher Scientific, Inc.). The membranes were
cross-linked in a UV transilluminator for 15 min and then were
incubated with blocking buffer and streptavidin-horseradish
peroxidase conjugates. Bound conjugates were detected using a
molecular imager (ChemiDoc XRS+; Bio-Rad Laboratories).
Chromatin immunoprecipitation assay
(ChIP)
EZ ChIP™ Chromatin Immunoprecipitation Kit (EMD
Millipore) was used to perform ChIP assays. In brief, cells were
crosslinked using 4% formaldehyde for 10 min followed by cell
collection and sonication with a predetermined power to yield
genomic DNA fragments of 200–1,000 bp. Antibodies (8 µg; Abcam)
were immobilized on Protein A/G magnetic beads (Life Technologies;
Thermo Fisher Scientific, Inc.) by overnight incubation. The
magnetic beads were washed to remove unconjugated antibody and
mixed with 8 µg of sonicated chromatin. After an overnight
incubation, magnetic beads were washed, and bound DNA was purified.
PCR was then performed using equal amounts of IN (input) and IP
(immunoprecipitated sample) DNA. The primers for PCR were as
follows: forward, 5′-GGAAAGAACCTTGTCTGCC-3′ and the reverse,
5′-GACTAAGTTCCTTTGAGGGC-3′. PCR products were analyzed by 15%
agarose gel electrophoresis.
Animal experiments
In vivo experiments were performed using 30
male nude mice (5–6 weeks old; 20–25 g body weight) purchased from
the Beijing Vital River Experimental Animals Co., Ltd. Mice were
housed in an air-conditioned atmosphere under a 12-h light/dark
cycle and given free access to food and water. The mice were
subcutaneously inoculated with 0.2 ml cell suspension
(2×106 cells, HepG2-SR/Huh7-SR or HepG2-SR/Huh7-SR
transfected with FoxM1 siRNA) using a sterile 22-gauge needle. When
tumors reached ~100 mm3, the mice were randomized into 6
groups with 5–8 mice per group. The groups with FoxM1 siRNA
transfection were treated with sorafenib (30 mg/kg/day orally) and
the other groups were treated with sorafenib or sorafenib combined
BEZ-235 (12.5 mg/kg/day orally). The tumor sizes and animal body
weights were measured twice weekly. After 1 week, tumors from a
single mouse from each group were dissected. Tumor tissues were
processed for immunoblotting as previously described (19). All animal experiments were performed
according to the protocol approved by the Army Medical University
Guidelines for Use and Care of Animals.
Statistical analysis
The data were expressed as the mean ± SD.
Comparisons between two groups were performed using one-way
analysis of variance (ANOVA) followed by Dunnett's test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Establishment of sorafenib-resistant
cell models
To investigate the mechanisms of sorafenib
resistance in liver cancer, HepG2 and Huh7 cells were chronically
exposed to sorafenib. Two different sorafenib-resistant cell lines
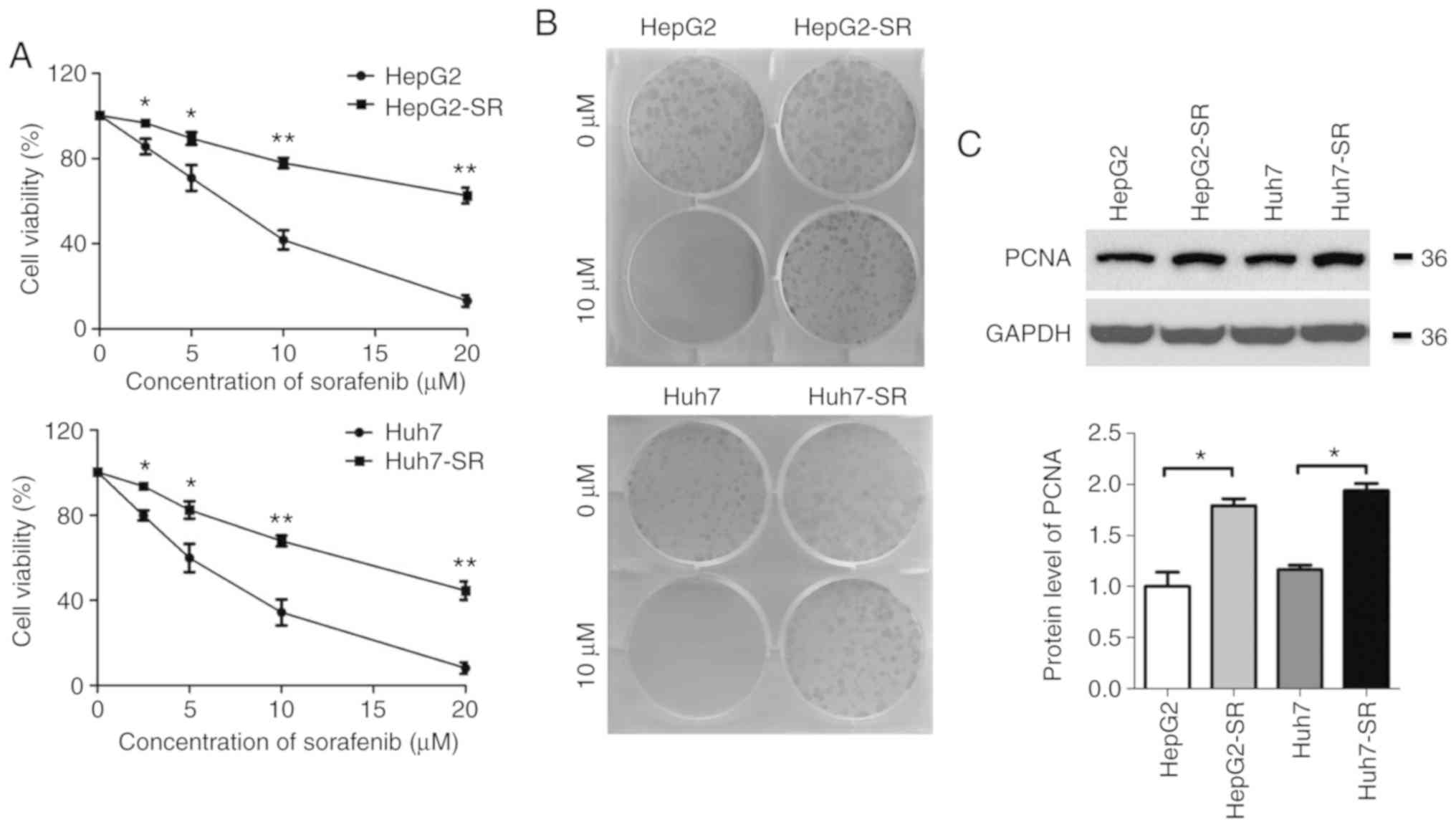
were established as previously described (20). As revealed in Fig. 1A, the IC50 value of
parental cells was ~6.2 µmol/l, whereas the IC50 was
>10 µmol/l in sorafenib-resistant cells. In the clonogenic
assay, no parental cell colonies were present after treatment with
10 µmol/l sorafenib for 1 week, whereas numerous
sorafenib-resistant cell colonies survived under the same
conditions (Fig. 1B). Additionally,
the levels of proliferating cell nuclear antigen (PCNA) were
significantly higher in sorafenib-resistant cells than in the
parental cells (Fig. 1C). These
results indicated that the sorafenib-resistant liver cancer cell
models had been established successfully.
FoxM1 is upregulated in
sorafenib-resistant liver cancer cells and involved in sorafenib
resistance
Since FoxM1 has an important role in drug
resistance, the expression of FoxM1 in liver cancer cells was
determined. Compared with the expression in parental cells, the
level of FoxM1 in sorafenib-resistant cells was significantly
increased, both at the protein and mRNA levels (Fig. 2A and B). Furthermore, knockdown of
FoxM1 significantly decreased the resistance of sorafenib in
sorafenib-resistant liver cancer cells (Fig. 2C and D). These data indicated that the
upregulation of FoxM1 has a key role in liver cancer resistance to
sorafenib.
AP1 enhances FoxM1 expression at the
transcriptional level in sorafenib-resistant liver cancer
cells
Since FoxM1 was upregulated at the mRNA and protein
levels, the activity of the FoxM1 promoter in sorafenib-resistant
liver cancer cells was examined. The FoxM1 promoter fragment was
cloned to construct a luciferase reporter plasmid. As revealed in
Fig. 3A, the activity of the FoxM1
promoter was significantly higher in sorafenib-resistant liver
cancer cells than in parental liver cancer cells. Furthermore,
bioinformatic analysis revealed that there is a transcription
factor AP1 binding site at −608 to −618 of the FoxM1 promoter
(Fig. 3B). A mutant luciferase
reporter plasmid was constructed in which the AP1 binding site in
the FoxM1 promoter was mutated (Fig.
3B). As revealed in Fig. 3C, the
promoter activity of the mutant luciferase reporter plasmid was
markedly lower than that of the wild-type luciferase reporter
plasmid in sorafenib-resistant liver cancer cells. Furthermore,
EMSA demonstrated that the wild-type AP1-binding sequence (GTT GAC
TAA GT), but not the mutant fragment (GCC TAC CGG GT), could bind
to the protein in nuclear extracts from sorafenib-resistant liver
cancer cells, and this binding could be blocked by the addition of
cold AP1 probe (Fig. 3D).
Furthermore, the ChIP assay revealed that the FoxM1 promoter
fragment could be amplified by PCR using c-jun
antibody-precipitated chromatin (Fig.
3E). These data demonstrated that AP1 enhanced the FoxM1
promoter activity in sorafenib-resistant liver cancer cells.
 | Figure 3.AP1 enhances FoxM1 expression at the
transcriptional level in sorafenib-resistant liver cancer cells.
(A) After transfection of HepG2/HepG2-SR and HuH-7/Huh7-SR cells
with pGL3-FoxM1 for 36 h, the luciferase activity was assayed using
the Dual-Luciferase Reporter System and normalized to the control.
**P<0.01. (B) Schematic representation of the FoxM1 promoter
region containing the putative binding sites for AP1. The region
(−2,000 to +100) containing the wild-type or mutant AP1 binding
site was cloned into the pGL3-Basic reporter vector and was named
pGL3-FoxM1 or pGL3-FoxM1-mut. (C) After transfection with
pGL3-FoxM1 or pGL3-FoxM1-mut for 36 h in HepG2-SR and Huh7-SR
cells, the luciferase activity was assayed using the
Dual-Luciferase Reporter System and normalized to the control.
**P<0.01. (D) The nuclear extracts from the HepG2 cells were
incubated with the indicated probes. Lane 1, labeled probe, nuclear
extracts, and 50-fold unlabeled probe; lane 2, labeled probe with
nuclear extracts; lane 3, labeled mutant probe and nuclear
extracts. The protein-DNA complexes formed are identified by
arrows. (E) Chromatin immunoprecipitation assay with chromatin
isolated from HepG2 cells. Antibody directed against c-Jun was used
for immune precipitation, using IgG as a negative control. Final
DNA extractions were amplified by PCR with the primer pairs
covering the binding site of AP1 in the FoxM1 promoter region.
Total extract (input) was used as a positive PCR control.
Representative data from three experiments are presented. The
experiment displayed is representative of at least three separate
experiments. AP1, activator protein-1; FoxM1, forkhead box M1; ns,
not significant. |
Knockdown of c-jun suppresses the
expression of FoxM1 and reduces the sorafenib resistance of liver
cancer cells
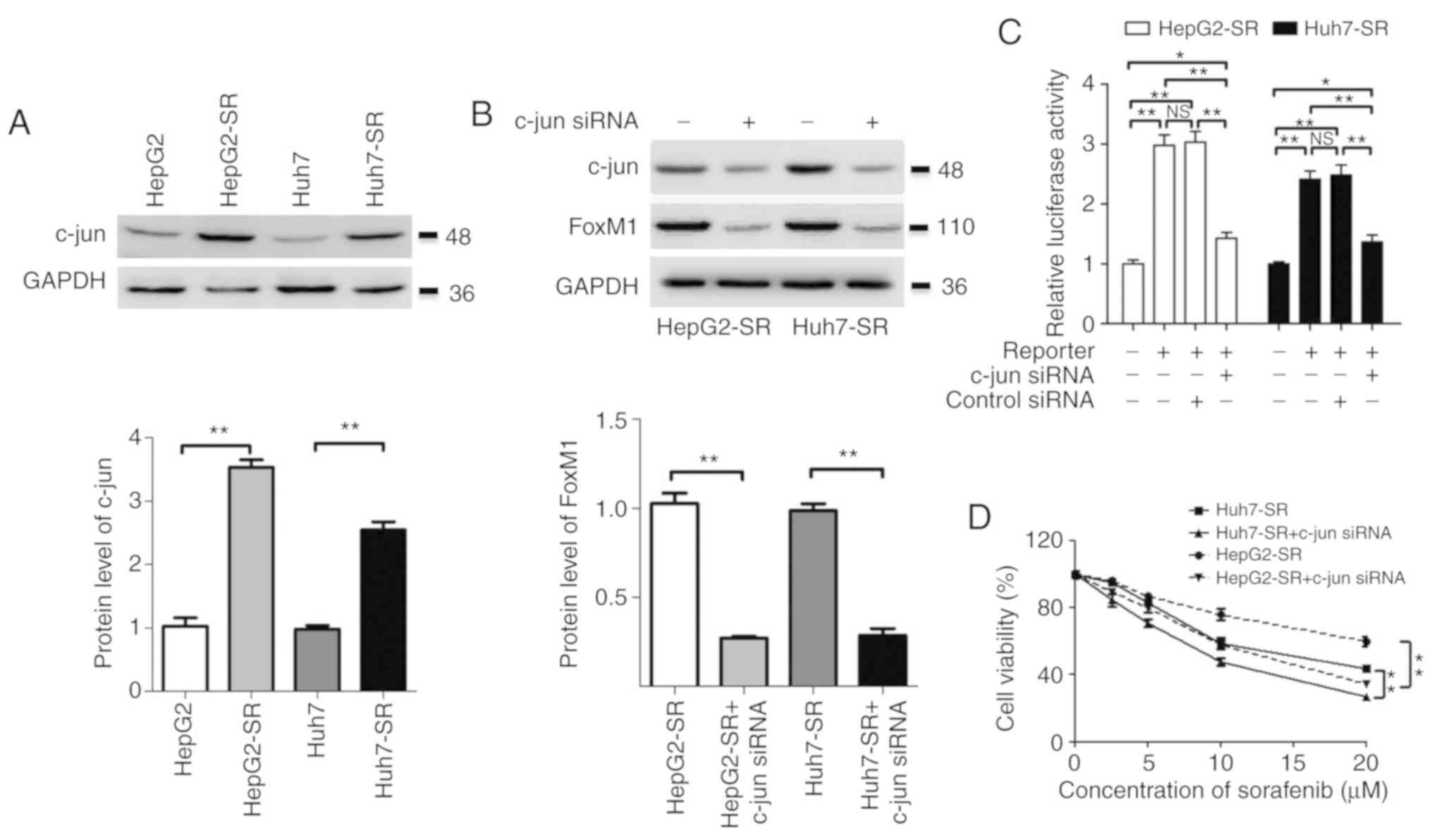
The expression of c-jun in liver cancer cells was
assessed. Compared to the expression in parental liver cancer
cells, the level of c-jun was markedly increased in
sorafenib-resistant liver cancer cells (Fig. 4A). Furthermore, knockdown of c-jun
significantly suppressed the expression of FoxM1 and downregulated
the promoter activity of FoxM1 in sorafenib-resistant liver cancer
cells (Fig. 4B and C). Moreover,
knockdown of c-jun also significantly reduced the resistance of
sorafenib-resistant liver cancer cells to sorafenib (Fig. 4D). These results indicated that the
overexpression of c-jun, at least partly, contributed to the
upregulation of FoxM1 and sorafenib resistance in liver cancer
cells.
Activation of AKT activates the
c-jun/FoxM1 signaling axis in sorafenib-resistant liver cancer
cells
The activity of several signaling pathways upstream
of c-jun was determined. Consistent with a previous study (3), ERK activity (the ratio of p-ERK to
t-ERK) was suppressed in sorafenib-resistant liver cancer cells
(Fig. 5A); however, the ratio of
p-AKT to t-AKT was significantly higher in sorafenib-resistant
liver cancer cells compared with parental cells (Fig. 5A), while there was no significant
difference in JNK activity (the ratio of p-JNK to t-JNK) between
the two groups (Fig. 5A). The AKT
inhibitor BEZ-235 significantly reduced the expression of c-jun and
FoxM1, and inhibited the activity of AKT (the ratio of p-AKT to
t-AKT), without an effect on JNK (the ratio of p-JNK to t-JNK) in
sorafenib-resistant liver cancer cells (Fig. 5B). However, the JNK inhibitor SP600125
inhibited the activity of JNK (the ratio of p-JNK to t-JNK), but
had no effect on the expression of c-jun and FoxM1 in sorafenib
resistance in liver cancer cells (Fig.
5C). The AKT inhibitor BEZ-235 also significantly reduced the
resistance of sorafenib-resistant liver cancer cells to sorafenib
(Fig. 5D). These results indicated
that the activation of AKT stimulated the c-jun/FoxM1 axis in
sorafenib-resistant liver cancer cells.
 | Figure 5.AKT activates the c-jun/FoxM1 axis in
sorafenib-resistant liver cancer cells. (A) The protein levels of
p-JNK, t-JNK, p-AKT, t-AKT, p-ERK and t-ERK were detected by
western blotting, and the ratios of p-JNK/t-JNK, p-AKT/t-AKT and
p-ERK/t-ERK were calculated after quantification using Quantity One
software in HepG2/HepG2-SR and HuH-7/Huh7-SR cells. (B) HepG2-SR
and Huh7-SR cells were treated with vehicle control or BEZ-235 (50
nM) for 24 h, then the protein levels of p-AKT, t-AKT, p-JNK,
t-JNK, c-Jun and FoxM1 were detected by western blotting, and the
ratios of p-JNK/t-JNK, p-AKT/t-AKT, c-Jun/GAPDH and FoxM1/GAPDH
were calculated after quantification using Quantity One software.
(C) HepG2-SR and Huh7-SR cells were treated with vehicle control or
SP600125 (10 µM) for 24 h, then the protein levels of p-JNK, t-JNK,
c-jun and FoxM1 were detected by western blotting, and the ratios
of p-JNK/t-JNK, c-Jun/GAPDH and FoxM1/GAPDH were calculated after
quantification using Quantity One software. (D) HepG2-SR and
Huh7-SR cells were treated with vehicle control or BEZ-235 (50 nM),
combined with sorafenib at increasing concentrations for 48 h. Then
the cell viability was measured by CCK-8 kit. The experiment
presented is representative of at least three independent
experiments. **P<0.01. FoxM1, forkhead box M1; p-,
phosphorylated-; ns, not significant. |
AKT inhibition and FoxM1 knockdown
increases sorafenib sensitivity of sorafenib-resistant liver cancer
cells in vivo
To investigate the effect of AKT inhibition and
FoxM1 knockdown on sorafenib resistance, sorafenib-resistant liver
cancer cells or FoxM1-knockdown sorafenib-resistant liver cancer
cells were implanted under the skin of nude mice. As revealed in
Fig. 6A and B, tumors derived from
sorafenib-resistant liver cancer cells were fast growing, even when
treated with sorafenib. While treatment with sorafenib combined
with BEZ-235, or sorafenib treatment together with FoxM1 knockdown
significantly delayed tumor growth (Fig.
6A and B). Western blot and qPCR analyses revealed that BEZ-235
and FoxM1 siRNA reduced the level of FoxM1 in tumor tissues, at
both the protein and mRNA levels (Fig. 6C
and D). BEZ-235 treatment also reduced the expression of c-jun
(Fig. 6C). These results demonstrated
that AKT inhibition or FoxM1 silencing increased the sorafenib
sensitivity of sorafenib-resistant liver cancer cells in
vivo.
Discussion
The clinical approval of sorafenib has been marked
as a new era in molecular targeted therapy for advanced liver
cancer (21). However, a phenomenon
termed ‘re-treatment response’ has been increasingly observed
(4). For example, certain patients
have a positive initial response to sorafenib treatment, but
treatment failure occurs over time. The acquired resistance of
cancer drugs remains a critical obstacle in cancer therapy. The
molecular mechanism of sorafenib resistance is still poorly
understood. The aim of the present study was to investigate the
molecular mechanisms of acquired resistance to sorafenib.
The forkhead box family consists of >50 mammalian
proteins, including FoxM1. FoxM1 is important for various
biological processes, such as proliferation, migration, metabolism
and invasion (15). As an oncogenic
transcription factor, FoxM1 expression is upregulated in various
types of cancer, including liver cancer (22). Previous studies have demonstrated that
FoxM1 is a key regulator of cell cycle progression. The activation
of FoxM1 has been revealed to be closely associated with the
activation of cell proliferation, invasion, migration and
tumorigenesis (23). Furthermore, the
deletion of FoxM1 results in cell cycle arrest and mitotic spindle
damage in vitro, and loss of FoxM1 leads to lethal embryonic
effects in vivo. FoxM1 is also an important molecular target
in drug-resistant tumors. Suppression of FoxM1 can sensitize tumor
cells to drug treatment (16,17). The combination of chemotherapeutic
regimens and FoxM1 inhibition may reduce treatment side effects,
and lead to the use of lower effective doses in patients. These
studies suggest that FoxM1 is an attractive target for liver cancer
therapy. Moreover, recent research has indicated that the
overexpression of FoxM1 predicts poor prognosis in patients with
liver cancer after resection (24). A
previous study revealed that sorafenib reduces the expression of
FoxM1 in liver cancer cells (25).
Our unpublished data also revealed that sorafenib treatment rapidly
downregulates FoxM1 in HepG2 and Huh7 cells; however, FoxM1 was
significantly upregulated in sorafenib-resistant liver cancer cells
treated with sorafenib for >1 month. These results indicated
that FoxM1 was downregulated initially, followed by upregulation as
liver cancer cells become resistant to sorafenib.
Multiple factors and various mechanisms regulate the
expression and activity of FoxM1. FoxM1 is regulated by microRNAs
(miRs), including miR-214 (26),
miR-197 (27), miR-34a (28), miR-361-5p (29), miR-761 (30) and miR-216b (31), and long non-coding RNAs, such as
LINC00339 (32), LOC653786 (19), ubiquitin-specific protease 5 (33), ubiquitin-conjugating enzyme E2C
(34) and ubiquitination E3-ligase
RNF168 (35). FoxM1 is
transcriptionally regulated by c-Myc (36), Her-2 (37), hypoxia-inducible factor (HIF)-1α and
HIF-2α (38); however, it is
currently unclear whether FoxM1 is regulated by AP1. The present
study demonstrated that FoxM1 upregulation mediated by AP1 promotes
sorafenib resistance in liver cancer cells. AP1 can bind to the
−608 to −618 site of the FoxM1 promoter and induce the
transcription of FoxM1.
The dimeric activator protein 1 (AP-1) transcription
factor family mainly consists of Jun (c-Jun, JunB and JunD) and Fos
proteins (c-Fos, FosB, Fra-1 and Fra-2) (39). c-Jun is expressed as an immediate
early gene in response to a variety of stress stimuli, growth
factors and subsequent signaling through MAP kinase pathways. It is
a major determinant of cell fate in the liver (40). Moreover, c-Jun acts as an oncogene in
the liver and strongly promotes liver tumorigenesis in models of
chemically-induced HCC (41).
Consistent with a previous study (42), the present study revealed that the
overexpression of c-Jun contributed to sorafenib resistance in
liver cancer cells. Generally, c-Jun is activated after
phosphrylation by JNK. Notably, in the present study it was
revealed that JNK inhibition did not change the level of c-Jun.
Since the levels of p-JNK between parental cells and
sorafenib-resistant cells did not exhibit significant differences,
thus it is surmised that the JNK/c-Jun pathway does not play a key
role in sorafenib-resistant cells.
The PI3K/AKT signaling pathway is involved in
hepatocarcinogenesis and chemoresistance in liver cancer cells
(43–45). The AKT inhibitor PI-103 was revealed
to enhance the inhibitory effects of sorafenib on cell
proliferation and tumorigenesis in liver cancer cells (46). The AKT inhibitor MK-2206 was revealed
to reverse the multidrug resistance and epithelial-mesenchymal
transition phenotype in sorafenib-resistant liver cancer cells
(44). Inhibition of the PI3K/AKT
signaling pathway using LY294002 can also reverse sorafenib-derived
chemoresistance in liver cancer (47). AKT pathways activate various
transcription factors, including AP1 (48). Consistent with the previous studies,
the findings of the present study demonstrated that the PI3K/AKT
signaling pathway was activated in sorafenib-resistant liver cancer
cells, followed by AP1 activation. The AKT/AP1/FoxM1 signaling axis
also contributed to sorafenib resistance. Suppression of this axis
by BEZ-235 significantly reduced sorafenib resistance in liver
cancer cells in vitro and in vivo.
In summary, the present study highlights an
important molecular mechanism of sorafenib resistance in liver
cancer cells. Targeting the AKT/AP1/FoxM1 signaling axis may
overcome sorafenib resistance in patients with liver cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81460558 and
81572375) and the Chongqing Natural Science Foundation
(cstc2018jcyjA2018).
Availability of data and materials
The datasets and certain material used and/or
analyzed during the present study are available from the
corresponding author on reasonable request.
Authors' contributions
JL and WC conceived and designed the experiments.
DY, XY and XD performed the experiments. JL and WC wrote the
manuscript. LC, LS, TL and FH ordered the reagents, collected the
materials and analyzed the data. All authors read and approved the
final manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All animal experiments were performed according to
the protocol approved by the Army Medical University Guidelines for
Use and Care of Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interests.
References
|
1
|
Khemlina G, Ikeda S and Kurzrock R: The
biology of Hepatocellular carcinoma: Implications for genomic and
immune therapies. Mol Cancer. 16:1492017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee DH and Lee JM: Primary malignant
tumours in the non-cirrhotic liver. Eur J Radiol. 95:349–361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eso Y and Marusawa H: Novel approaches for
molecular targeted therapy against hepatocellular carcinoma.
Hepatol Res. 48:597–607. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ray EM and Sanoff HK: Optimal therapy for
patients with hepatocellular carcinoma and resistance or
intolerance to sorafenib: Challenges and solutions. J Hepatocell
Carcinoma. 4:131–138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao S, Fan LY and Lam EW: The FOXO3-FOXM1
axis: A key cancer drug target and a modulator of cancer drug
resistance. Semin Cancer Biol. 50:77–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frau M, Feo F and Pascale RM: Pleiotropic
effects of methionine adenosyltransferases deregulation as
determinants of liver cancer progression and prognosis. J Hepatol.
59:830–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang IC, Chen YJ, Hughes D, Petrovic V,
Major ML, Park HJ, Tan Y, Ackerson T and Costa RH: Forkhead box M1
regulates the transcriptional network of genes essential for
mitotic progression and genes encoding the SCF (Skp2-Cks1)
ubiquitin ligase. Mol Cell Biol. 25:10875–10894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Z, Ahmad A, Li Y, Banerjee S, Kong D
and Sarkar FH: Forkhead box M1 transcription factor: A novel target
for cancer therapy. Cancer Treat Rev. 36:151–156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Laoukili J, Stahl M and Medema RH: FoxM1:
At the crossroads of ageing and cancer. Biochim Biophys Acta.
1775:92–102. 2007.PubMed/NCBI
|
|
10
|
Ma RY, Tong TH, Leung WY and Yao KM:
Raf/MEK/MAPK signaling stimulates the nuclear translocation and
transactivating activity of FOXM1. Methods Mol Biol. 647:113–123.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Balli D, Zhang Y, Snyder J, Kalinichenko
VV and Kalin TV: Endothelial cell-specific deletion of
transcription factor FoxM1 increases urethane-induced lung
carcinogenesis. Cancer Res. 71:40–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Zhang N, Dai B, Liu M, Sawaya R,
Xie K and Huang S: FoxM1B transcriptionally regulates vascular
endothelial growth factor expression and promotes the angiogenesis
and growth of glioma cells. Cancer Res. 68:8733–8742. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Banerjee S, Kong D, Li Y and
Sarkar FH: Down-regulation of Forkhead Box M1 transcription factor
leads to the inhibition of invasion and angiogenesis of pancreatic
cancer cells. Cancer Res. 67:8293–8300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan Y, Raychaudhuri P and Costa RH: Chk2
mediates stabilization of the FoxM1 transcription factor to
stimulate expression of DNA repair genes. Mol Cell Biol.
27:1007–1016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Myatt SS and Lam EW: The emerging roles of
forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kwok JM, Peck B, Monteiro LJ, Schwenen HD,
Millour J, Coombes RC, Myatt SS and Lam EW: FOXM1 confers acquired
cisplatin resistance in breast cancer cells. Mol Cancer Res.
8:24–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carr JR, Park HJ, Wang Z, Kiefer MM and
Raychaudhuri P: FoxM1 mediates resistance to herceptin and
paclitaxel. Cancer Res. 70:5054–5063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sebaugh JL: Guidelines for accurate
EC50/IC50 estimation. Pharm Stat. 10:128–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang F, Wu Q, Zhang Y, Xiong H, Li X, Li
B, Xie W, Zhang L, Xu M, Zhang K and He F: LncRNA LOC653786
promotes growth of RCC cells via upregulating FOXM1. Oncotarget.
9:12101–12111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
da Motta Girardi D, Correa TS, Crosara
Teixeira M and Dos Santos Fernandes G: Hepatocellular carcinoma:
Review of targeted and immune therapies. J Gastrointest Cancer.
49:227–236. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu QF, Liu C, Tai MH, Liu D, Lei L, Wang
RT, Tian M and Lü Y: Knockdown of FoxM1 by siRNA interference
decreases cell proliferation, induces cell cycle arrest and
inhibits cell invasion in MHCC-97H cells in vitro. Acta Pharmacol
Sin. 31:361–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qu K, Xu X, Liu C, Wu Q, Wei J, Meng F,
Zhou L, Wang Z, Lei L and Liu P: Negative regulation of
transcription factor FoxM1 by p53 enhances oxaliplatin-induced
senescence in hepatocellular carcinoma. Cancer Lett. 331:105–114.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun H, Teng M, Liu J, Jin D, Wu J, Yan D,
Fan J, Qin X, Tang H and Peng Z: FOXM1 expression predicts the
prognosis in hepatocellular carcinoma patients after orthotopic
liver transplantation combined with the Milan criteria. Cancer
Lett. 306:214–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei JC, Meng FD, Qu K, Wang ZX, Wu QF,
Zhang LQ, Pang Q and Liu C: Sorafenib inhibits proliferation and
invasion of human hepatocellular carcinoma cells via up-regulation
of p53 and suppressing FoxM1. Acta Pharmacol Sin. 36:241–251. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian C, Wu H, Li C, Tian X, Sun Y, Liu E,
Liao X and Song W: Downreguation of FoxM1 by miR-214 inhibits
proliferation and migration in hepatocellular carcinoma. Gene Ther.
25:312–319. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu Q, Du K, Mao X and Ning S: miR-197 is
downregulated in cervical carcinogenesis and suppresses cell
proliferation and invasion through targeting forkhead box M1. Oncol
Lett. 15:10063–10069. 2018.PubMed/NCBI
|
|
28
|
Segal NH, He AR, Doi T, Levy R, Bhatia S,
Pishvaian MJ, Cesari R, Chen Y, Davis CB, Huang B, et al: Phase I
study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137
agonist, in patients with advanced cancer. Clin Cancer Res.
24:1816–1823. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian L, Zhao Z, Xie L and Zhu J:
MiR-361-5p suppresses chemoresistance of gastric cancer cells by
targeting FOXM1 via the PI3K/Akt/mTOR pathway. Oncotarget.
9:4886–4896. 2017.PubMed/NCBI
|
|
30
|
Cao S, Lin L, Xia X and Wu H: MicroRNA-761
promotes the sensitivity of colorectal cancer cells to
5-Fluorouracil through targeting FOXM1. Oncotarget. 9:321–331.
2017.PubMed/NCBI
|
|
31
|
He S, Liao B, Deng Y, Su C, Tuo J, Liu J,
Yao S and Xu L: MiR-216b inhibits cell proliferation by targeting
FOXM1 in cervical cancer cells and is associated with better
prognosis. BMC Cancer. 17:6732017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yuan Y, Haiying G, Zhuo L, Ying L and Xin
H: Long non-coding RNA LINC00339 facilitates the tumorigenesis of
non-small cell lung cancer by sponging miR-145 through targeting
FOXM1. Biomed Pharmacother. 105:707–713. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li XY, Wu HY, Mao XF, Jiang LX and Wang
YX: USP5 promotes tumorigenesis and progression of pancreatic
cancer by stabilizing FoxM1 protein. Biochem Biophys Res Commun.
492:48–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo L, Ding Z, Huang N, Huang Z, Zhang N
and Xia Z: Forkhead Box M1 positively regulates UBE2C and protects
glioma cells from autophagic death. Cell Cycle. 16:1705–1718. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kongsema M, Zona S, Karunarathna U,
Cabrera E, Man EP, Yao S, Shibakawa A, Khoo US, Medema RH, Freire R
and Lam EW: RNF168 cooperates with RNF8 to mediate FOXM1
ubiquitination and degradation in breast cancer epirubicin
treatment. Oncogenesis. 5:e2522016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pan H, Zhu Y, Wei W, Shao S and Rui X:
Transcription factor FoxM1 is the downstream target of c-Myc and
contributes to the development of prostate cancer. World J Surg
Oncol. 16:592018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qi W, Li X, Zhang Y, Yao R, Qiu W, Tang D
and Liang J: Overexpression of Her-2 upregulates FoxM1 in gastric
cancer. Int J Mol Med. 33:1531–1538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bai C, Liu X, Qiu C and Zheng J: FoxM1 is
regulated by both HIF-1α and HIF-2α and contributes to
gastrointestinal stromal tumor progression. Gastric Cancer.
22:91–103. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Eferl R and Wagner EF: AP-1: A
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fuest M, Willim K, Macnelly S, Fellner N,
Resch GP, Blum HE and Hasselblatt P: The transcription factor c-Jun
protects against sustained hepatic endoplasmic reticulum stress
thereby promoting hepatocyte survival. Hepatology. 55:408–418.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen
X, Chen L, Scheuch H, Zheng H, Qin L, et al: Liver cancer
initiation is controlled by AP-1 through SIRT6-dependent inhibition
of survivin. Nat Cell Biol. 14:1203–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Haga Y, Kanda T, Nakamura M, Nakamoto S,
Sasaki R, Takahashi K, Wu S and Yokosuka O: Overexpression of c-Jun
contributes to sorafenib resistance in human hepatoma cell lines.
PLoS One. 12:e01741532017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kunter I, Erdal E, Nart D, Yilmaz F,
Karademir S, Sagol O and Atabey N: Active form of AKT controls cell
proliferation and response to apoptosis in hepatocellular
carcinoma. Oncol Rep. 31:573–580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dong J, Zhai B, Sun W, Hu F, Cheng H and
Xu J: Activation of phosphatidylinositol 3-kinase/AKT/snail
signaling pathway contributes to epithelial-mesenchymal
transition-induced multi-drug resistance to sorafenib in
hepatocellular carcinoma cells. PLoS One. 12:e01850882017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang PF, Li KS, Shen YH, Gao PT, Dong ZR,
Cai JB, Zhang C, Huang XY, Tian MX, Hu ZQ, et al: Galectin-1
induces hepatocellular carcinoma EMT and sorafenib resistance by
activating FAK/PI3K/AKT signaling. Cell Death Dis. 7:e22012016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gedaly R, Angulo P, Chen C, Creasy KT,
Spear BT, Hundley J, Daily MF, Shah M and Evers BM: The role of
PI3K/mTOR inhibition in combination with sorafenib in
hepatocellular carcinoma treatment. Anticancer Res. 32:2531–2536.
2012.PubMed/NCBI
|
|
47
|
Zhang H, Wang Q, Liu J and Cao H:
Inhibition of the PI3K/Akt signaling pathway reverses
sorafenib-derived chemo-resistance in hepatocellular carcinoma.
Oncol Lett. 15:9377–9384. 2018.PubMed/NCBI
|
|
48
|
Kumar D, Tewari-Singh N, Agarwal C, Jain
AK, Inturi S, Kant R, White CW and Agarwal R: Nitrogen mustard
exposure of murine skin induces DNA damage, oxidative stress and
activation of MAPK/Akt-AP1 pathway leading to induction of
inflammatory and proteolytic mediators. Toxicol Lett. 235:161–71.
2015. View Article : Google Scholar : PubMed/NCBI
|