Introduction
A total of 3 forms of arsenic compounds have been
characterized, including inorganic arsenic, organic arsenic and
arsenic gas. Inorganic arsenic is metabolized by a sequential
process to organic arsenic in mammals (1). Arsenic and inorganic arsenic compounds
are classified into Group 1 carcinogens for humans by the
International Agency for Research on Cancer (IARC) (2). Arsenic is derived from volcanic and
industrial activities and is emitted to the air, water and soil.
The anthropogenic contamination of arsenic includes mining, burning
of fossil fuels, smelting of non-ferrous metals, agriculture
pesticides and timber preservative agents (2). The ingestion of contaminated water and
food is the major route of arsenic exposure for the general
population compared with air pollution.
The carcinogenicity of arsenic in humans is
concluded by several epidemiological studies. Induction of lung
cancer was reported by inhalation of arsenic occurring in copper
smelters (3). Oral exposure of
arsenic can cause skin (4,5), bladder (6,7), lung
(8) and liver cancers (9). In laboratory rodent studies, oral
arsenic induced cancer at a very high dose compared with the levels
of exposure in the human population (10). In male F344 rats, dimethylarsinic acid
induced urothelial carcinoma at a dosage higher than 50 ppm
following 104 weeks of treatment (11). An additional study reported that
dimethylarsinic acid induced urothelial carcinogenesis in rats
(12) but not in mice (13). The dose of exposure used in mice was
500 ppm and the exposure period was 2 years (13). Although tumor formation in mice was
not present, treatment of C57BL/6 mice with inorganic arsenic for 6
days at 50 ppm caused urothelial hyperplasia (14). Using scanning electronic microscopy
(SEM), a previous study indicated that sodium arsenic could induce
urothelial hyperproliferation, indicative of hyperplasia following
7 days of arsenic exposure at 25 ppm (+3 oxidation state) in
methyltransferase knockout mice (15). This finding indicated that arsenic
induced urothelial cytotoxicity. In addition, arsenic and the
carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine have
been revealed to mutually promote mouse urothelial carcinogenesis
(16). These studies indicated that
the interaction of arsenic with other environmental carcinogens may
be an important cause for arsenic-induced bladder cancer.
Despite the hazardous effects of arsenic in humans
and laboratory animals, the underlining mechanism of
arsenic-induced bladder damage has not been fully explained due to
its complexity. In terms of gene regulation, several studies have
been conducted using cDNA array analysis for investigating
arsenic-induced changes in gene expression in vitro. For
example, E-cadherin expression was revealed to be enhanced
and integrin β3 expression decreased by sodium arsenite
(iAsIII), monomethylarsonous acid (MMAIII),
and dimethylarsinous acid (DMAIII) treatment in SV-HUC-1
immortalized human uroepithelial cells (17). Chronic 12-week exposure to 50 nM of
MMAIII led to substantial biological and functional gene
expression changes in another immortalized human uroepithelial cell
line, namely UROtsa (18).
In the present study, an in vivo mouse model
was initially used to analyze the global gene expression changes
induced by arsenic, and the association of gene expression and DNA
methylation was further assayed in certain cancer-related genes. In
addition, the selected genes were further examined in human
urothelial cells in vitro. The results indicated that
arsenic-induced methylation inhibition of human WIF1 gene,
and human WIF1 gene is always methylation inhibited in
bladder cancer. It provides useful information that WIF1
gene expression may be a biomarker for bladder cancer.
Materials and methods
Animals and arsenic treatment
Female (n=20 each) 8-week-old C57BL/6 mice were
purchased from the National Laboratory Animal Center (Taipei,
Taiwan). All animals were maintained at the qualified animal care
facility of The Biotechnology and Health Hall of National Chiayi
University for a 1-week of adaptation. All mice were housed in
polycarbonate cages, provided with food and water ad libitum
and maintained on a 12-h light-dark cycle at 22±2°C with 60±5%
humidity. At 9 weeks of age, mice were administrated 50 ppm arsenic
(0.0867 mg/ml sodium arsenite, Sigma-Aldrich; Merck KGaA) (arsenic
group) in drinking water or water only (control group) for 2 weeks.
The drinking water was renewed weekly and mouse body weights were
recorded twice weekly. The mice were monitored daily for their
health condition by observing specific behaviors including squeals,
decreased locomotor activity, and abnormal posture. The humane
endpoint was reached when the weight loss of each mouse was
>20%. At the endpoint, each mouse was placed in a transparent
polycarbonate cage and sacrificed by gradual-fill CO2
exposure with a displacement rate of 20% chamber volume/min. When
unconsciousness and breathing arrest were observed, the
CO2 flow was maintained for about 1 min. The death of
each mouse was confirmed by prolonged palpation of heartbeat stop
for 30 sec (19) accompanied by no
response observed to toe pinch reflex. Next, cervical dislocation
was performed to assure euthanasia (20) and then the bladder was collected. The
animal experiments were approved by the Institutional Animal Care
and Use Committee of National Chiayi University approval no.
103040.1 and according to the guidelines of The Animal Research:
Reporting in vivo Experiments, recommended by The National
Centre for the Replacement, Refinement and Reduction of Animals in
Research (21).
Bladder tissue collection for
hematoxylin and eosin (H&E) staining
Following 2 weeks of treatment, the mice were
euthanized and the bladder tissues were collected. A total of 12
bladders (6/group) were fixed in formaldehyde, dehydration and then
embedded in paraffin. The embedded tissues were cut into 3-µm
sections on glass slides and then were stained with H&E for
histopathologic examination (empty bladder).
Bladder tissue collection for SEM
examination
A total of 12 bladders (6/group) were used in this
experiment. Following 2 weeks of treatment, the mice were
euthanized and the bladder tissues were inflated in situ
with Bouin's fixative (150–200 µl/bladder), and after being tied
with string and removal, the bladders were placed in Bouin's
fixative for 1 h. Following fixation, the Bouin's-fixed bladders
were cut in half longitudinally. One-half of a bladder was used for
SEM examination and the other was used for H&E staining
(inflated bladder).
Cell culture condition
Human urinary tract epithelial cell line SV-HUC-1
and human bladder carcinoma cell lines 5637, RT4, HT1376, T24,
TSGH8301 and BFTC905 were purchased from Bioresource Collection and
Research Center (BCRC), and J82 was obtained from ATCC. SV-HUC-1
was cultured in Ham's F12 medium with 7% fetal bovine serum (FBS);
RT4 and T24 were cultured in McCoys 5a medium with 10% FBS; J82 and
HT1376 were cultured in MEM medium with 10% FBS; 5637, TSGH8301 and
BFTC905 were cultured in RPMI-1640 medium with 10% FBS. All culture
mediums contained 100 U/ml penicillin and 100 µg/ml streptomycin.
Cells were inculated in a CO2 incubator at 37°C, with 5%
CO2 and 95% filtered air.
Extraction of genomic DNA from mouse
bladders and a human urinary epithelial cell line
After euthanization, the mouse bladders (n=2 each)
were isolated and excised for exposure of the inner urothelium.
After adding cell lysis buffer (10 mM Tris-Cl, pH 8.0, 1 mM EDTA,
pH 8.0, 0.1% SDS) to the tube containing the excised bladder, the
bladder was gently homogenized with a plastic pestle. The
non-homogenized tissue was discarded and proteinase K (0.1 mg/ml)
was added, and then incubation followed at 55°C for 16 h.
DNase-free RNase (0.02 mg/ml) was added at 37°C for 1 h to remove
RNA, then potassium acetate solution was added and mixed by
inverting the tube. After centrifuging at 13,200 × g, at 4°C for 10
min, the clear supernatant was collected. Genomic DNA was
precipitated by adding isopropanol and centrifuged at 13,200 × g,
at 4°C for 1 min. The DNA pellet was washed twice by cold 70%
ethanol, and then dissolved in an appropriate volume of TE buffer
(pH 7.6). SV-HUC-1 cells were treated with or without 0.5 µM sodium
arsenite for 2 and 10 weeks. After washing the cells by PBS (137 mM
NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM
KH2PO4, pH 7.4) and lysing the cells using
cell lysis buffer, the genomic DNA was collected according to the
same process as aforementioned.
Extraction of RNA from mouse bladders
and a human urinary epithelial cell line
The mouse bladders (n=6 each) were isolated and sunk
in RNAlater (Invitrogen; Thermo Fisher Scientific, Inc.) at 4°C for
24 h. The bladders were transferred to TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and were homogenized by a plastic
pestle. The non-homogenized tissue was discarded, and the total RNA
was extracted according to the manufacturer's instructions. For
SV-HUC-1 cells, after washing the cells by PBS and lysing cells
using TRIzol reagent, the RNA was collected according to the same
process as aforementioned. The concentration and purity of the RNA
was measured by a NanoDrop 1000 spectrophotometer. Purity was
verified using the ratio of the OD260/OD280
and OD260/OD230. For the microarray assay,
the quality of the mouse total RNA was further accessed by Agilent
2100 Bioanalyzer.
Analysis of mRNA expression
alternation using mouse oligonucleotide DNA microarray chip
One µg RNA was obtained from the total RNA of each
mouse bladder (6 in the control and 6 in the arsenic group) to pool
together to produce 2 mixed RNA samples (1 control and 1 arsenic
group). Fluorescent aRNA targets were prepared from 1 µg of the
mixed RNA samples using OneArray® Amino Allyl aRNA
Amplification kit (Phalanx Biotech Group) and Cy5 dye (GE
Healthcare Life Sciences). Fluorescent targets were hybridized to
the Mouse OneArray® (Phalanx Biotech Group) with Phalanx
hybridization buffer using Phalanx Hybridization system. After 16 h
of hybridization, non-specific binding targets were removed using
saline and sodium citrate buffer. The slides were scanned using a
DNA Microarray Scanner (Model G2505C; Agilent Technologies, Inc.).
The Cy5 fluorescent intensities of each spot were analyzed using
GenePix 4.1 software (Molecular Devices, LLC). Each single sample
was assessed at least twice in terms of technical or biological
replicates with a reproducibility >0.975. The signal intensity
of each spot was loaded into the Rosetta Resolver
System® (Rosetta Biosoftware) to process the data
(22). The error model of the Rosetta
Resolver System® could remove both systematic and random
errors from the data. The spots that were flagged to be <0 were
filtered out. Spots that passed the criteria were normalized by 50%
median scaling normalization method. The technical repeat data was
tested by Pearson's correlation coefficient calculation to verify
the reproducibility (R value >0.975). Normalized spot
intensities were transformed to gene expression log2
ratios between the control and treatment groups. The fold change
and P-value for pair-wise sample comparisons were calculated to
evaluate differentially expressed genes (DEGs). The criteria with
log2|fold change|≥0.58 and P<0.05 were used for
further analysis. The log2|fold change|≥0.58 is an
acceptable value for a microarray study (23).
Reverse transcription-quantitative
real-time polymerase chain reaction (RT-PCR) analysis
Four mouse genes, including adenosine A1 receptor
(Adora1), cystathionine beta-synthase (Cbs),
metastasis associated lung adenocarcinoma transcript 1
(Malat1), and Wnt inhibitory factor 1 (Wif1), were
selectively targeted. Each reaction included 20 ng cDNA, 500 nM
forward and reverse primers, and 2X Fast SYBR-Green PCR Master mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.). A total of 10
µl reaction volumes were used for RT-PCR with the specific primers
listed as follows: Mouse Adora1 forward,
5′-GAGGCGGACATCACATTCCAT-3′ and reverse,
5′-AGCCACCTCACTCACCCTAGA-3′; Mouse Cbs forward,
5′-GCTGAACCAGACGGAGCAAA-3′ and reverse, 5′-GGGCAAAGGCGAAGGAATCT-3′;
Mouse Malat1 forward, 5′-AGGGTTCTAAAGGCTCTGGGTA-3′ and
reverse, 5′-AAGACGAATTGGGCATAACCTGAA-3′; Mouse Wif1 forward,
5′-TTTGTGGTCTTAGAATGGGGAGTG-3′ and reverse,
5′-ACGCTGCTATTGGCTTTATCCG-3′. Each sample was assessed in
triplicate. The Bio-Rad CFX Connect real-time PCR instrument and
the Bio-Rad CFX Manager version 3.0 software (both from Bio-Rad
Laboratories, Inc.) were used for the experimental setup and data
analysis. The RT-PCR data of the target genes were normalized
against the reference gene GAPDH by using its specific
primers forward, 5′-AAGGTCGGTGTGAACGGATT-3′ and reverse,
5′-GTGAGTGGAGTCATACTGGAACAT-3′.
Bisulfite conversion of genomic DNA
and analysis of DNA methylation levels in mouse bladder tissue and
human urinary epithelial cells
Five hundred nanograms of genomic DNA was subjected
to sodium bisulfite modification using the EZ DNA methylation-Gold™
kit (Zymo Research Corp.). Mouse DNA methylation levels were
analyzed by bisulfite-sequencing PCR (BSP) method using bisulfite
specific primers. The BSP primers for the mouse Adora1 gene
were 5′-TTTGAGTTTYGTAGGTGATTAGGGTTTGGGTTG-3′ (forward) and
5′-CTAACCACCTAAACTATCTAACCAAATATCCCC-3′ (reverse), which amplified
−282 to 201 bp of the mouse Adora1 gene (483 bp); for the
mouse Cbs gene they were
5′-AAAYGAGGTTTTTTGATATTTAGTTAGGTTGTG-3′ (forward) and
5′-CRTCCAAATACAAAAAAAACCAAATCC-3′ (reverse), which amplified −298
to 271 bp of the mouse Cbs gene (569 bp). Next, the PCR
products were subcloned into the T&A cloning vector (Yearstern
Biotech). To determine the CpG methylation level of the 5′CpG
island of each gene, 10 clones of each gene were randomly selected
for sequencing. The BSP primers for the human WIF1 gene were
5′-TAGGGGTTTTTGAGTGTTT-3′ (forward) and
5′-ACCTAAATACCAAAAAACCTAC-3′ (reverse), which amplified −168 to 236
bp of the human WIF1 gene (404 bp). Then, a second round of
nested methylation-specific PCR (MSP) or unmethylation-specific PCR
(USP) was performed using the BSP-PCR products as templates. The
MSP for the human WIF1 gene were 5′-CGTTTTATTGGGCGTATCGT-3′
(forward) and 5′-ACTAACGCGAACGAAATACGA-3′ (reverse), 145 bp, and
for the USP they were 5′-GGGTGTTTTATTGGGTGTATTGT-3′ (forward) and
5′-AAAAAAACTAACACAAACAAAATACAAAC-3′ (reverse), 154 bp. The MSP and
USP PCR products were analyzed by 1% agarose gel. No regular
loading control such as a housekeeping gene for the MSP/USP assay
was used since its aim was to determine the relative methylation
status of the WIF1 gene instead of the absolute expression
level. A subsequent RT-PCR was applied to confirm the mRNA level of
WIF1 under such methylation status with GAPDH as the
loading control.
RNA expression detection of human
ADORA1, CBS, MALAT1 and WIF1 genes in human urinary epithelial
cells
Total RNA was isolated from cells. Reverse
transcription (RT) was performed on 2 µg of total RNA by 1.5 µM
random hexamer and RevertAid™ reverse transcriptase (Fermentas;
Thermo Fisher Scientific, Inc.). Then, 1/20 volume of reaction
mixture was used for PCR with human ADORA1-specific primers
(5′-TTTGGTGACCTTGGGTGCTT-3′, 5′-ACCACCATCTTGTACCGGAG-3′, product
size 427 bp); CBS-specific primers
(5′-GTCGCTCAGGAACTTGGTCA-3′, 5′-GAACCAGACGGAGCAGACAA-3′, product
size 286 bp); MALAT1-specific primers
(5′-TTATCCAGTGACTAAAACCAAC-3′, 5′-AAAAGGAGAAATACAGAAAGAG-3′,
product size 443 bp); WIF1-specific primers
(5′-CACTCGCAGATGCGTCTTTC-3′, 5′-CCAACCGTCAATGTCCCTCT-3′, product
size 251 bp); and GAPDH-specific primers
(5′-CAAGGTCATCCATGACAACTTTG-3′, 5′-GTCCACCACCCTGTTGCTGTAG-3′,
product size 496 bp). The PCR products were analyzed by 1–2%
agarose gel.
Cell proliferation assay by
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT)
The number of proliferating cells was determined by
a colorimetric MTT assay. After cells were treated with or without
0.5 µM sodium arsenite for 14 days, the cells were seeded in
96-well plates without sodium arsenite, then were incubated for 2 h
(Day 0) and for 1–4 days. MTT was added into the medium for 2 h,
then the medium was discarded and dimethyl sulfoxide was added to
dissolve the formazan product. Each well was measured by light
absorbance at 490 nm. The result was expressed as the fold to the
Day 0 control group.
Cell migration analysis by
Transwell
A cell migration assay was conducted using 24-well
Transwell inserts (8 µm pore PET filters; EMD Millipore). In
brief, after arsenic treatment, SV-HUC-1 cells (2×105
cells/well) were placed in the upper chamber of the Transwell
insert in serum-free medium, and then incubated for 24 h. Medium
containing 7% FBS was placed in the lower chamber. At the end of
the incubation period, the non-migrated cells were removed using a
cotton swab; the migrated cells maintained in the insert filter
were fixed with 4% formaldehyde (at 25°C for 5 min) and stained
with 0.5% crystal violet (25°C, 5 min). In the lower surface of the
filter, cells penetrated were counted and photographed under a
phase-contract microscope at a magnification of ×100. The crystal
violet was dissolved by 100 µl DMSO/insert and counted by a
spectrophotometer at 580 nm. Three independent experiments were
performed.
Statistical analysis
The statistical analysis was performed using
SigmaPlot version 12.5 (Systat Software, Inc.). Statistical
differences were analyzed by Student's t-test and a P-value
<0.05 was considered to indicate a statistically significant
difference. Microarray data were processed using the Rosetta
Resolver System®, which performs unique error modeling
to adjust for background noise and fractional noise. The P-value
representing the probability of differentially expressed genes was
thereby generated by a patented system according to a standard
Gaussian distribution based on standardized variance of the
intensity difference (22).
Results
Short-term treatment of high-dose
sodium arsenite induces hyperplasia and dysplasia in mouse
urothelium
It has been revealed that a clear morphological
change in the mouse urothelium can occur following 50 ppm of
arsenic treatment for 6 days (14).
Therefore, in the present study, a high-dose (50 ppm) of arsenic
treatment was used for a short-term period (14 days). The study
aimed to identify the morphological changes induced by arsenic in
urothelial cells and the corresponding alterations in gene
expression. Following 50 ppm of arsenic intake for 2 weeks, the
mice were alive and their body weight was gradually increased
(Fig. 1A). No significant difference
was noted in the body weight change between the control and the
arsenic-treated animal groups(Fig.
1A). This suggested that 50 ppm of arsenic intake that was
administered for a 2-week duration may not significantly affect the
animal appetite or their general health condition. The exterior
appearance and physical activity of the animals further indicated
no noticeable difference between the control and the
arsenic-treated groups. Following euthanasia, the mouse bladders
were harvested for histopathological and genetic assessments. The
histopathological examination (Fig. 1B
and C) revealed that arsenic induced 83.3% (10/12) hyperplasia
and 8.3% (1/12) dysplasia in mouse urothelium, which represented
the precancerous characteristics of the urothelium. In the
hyperplastic tissue, the analysis indicated increased thickness of
the urothelium for more than four layers without cellular atypia
(Fig. 1C, arsenic group). In the
dysplastic tissue, the analysis indicated apparent cytologic and
architectural atypia of the urothelium without evident invasion.
Moreover, intracellular inclusions were present within the umbrella
cells of the sodium arsenite-treated mice (Fig. 1C, arrow), which acted as a detoxifying
sequestration mechanism (24). Using
SEM, the accumulation of round cells was observed, which further
indicated urothelial hyperplasia (Fig.
1D, arsenic group). Certain surface leakages were also present
in arsenic-treated mice (Fig. 1D).
Furthermore, arsenic caused additional extensive hydropic
degeneration of hepatocytes, characterized by diffuse vacuolated
swelling of the hepatocytic cytoplasm and narrowed sinusoidal
capillary spaces (Fig. S1).
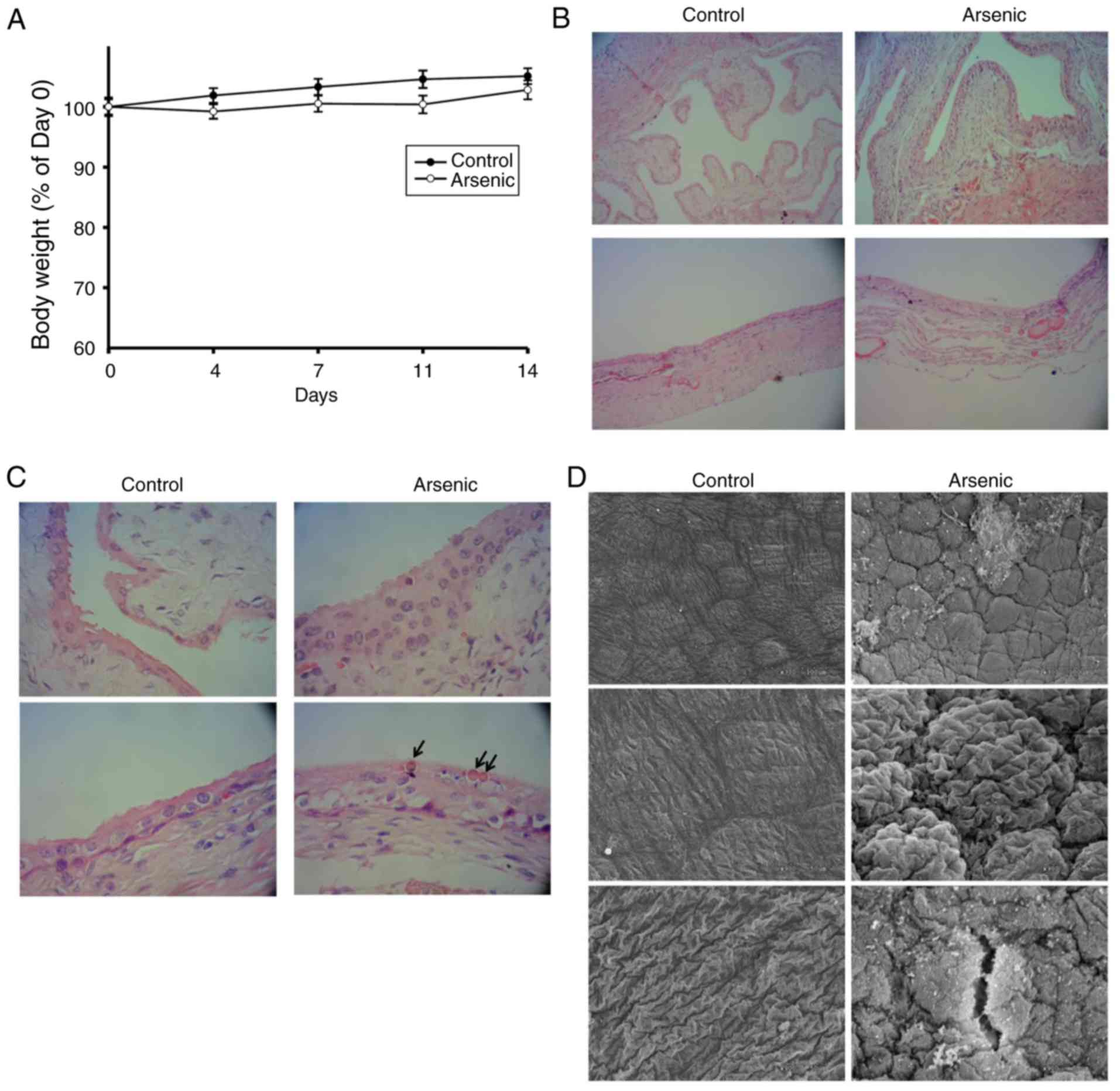 | Figure 1.The body weight change and bladder
morphologic alternation of the mice. Mice were administrated 0 ppm
(Control, n=12) and 50 ppm arsenic (Arsenic, n=12) in drinking
water, respectively, for 14 days. (A) They were weighed 1 day
before Day 1 (Day 0), and Days 4, 7, 11 and 14. The weight of Day 0
was set as 100%. After euthanization, the bladders were harvested
for H&E staining and examined by light microscopy at a
magnification of (B) ×100 and (C) ×400 magnification. Upper panels:
Empty bladder (n=6 in each group), lower panels: Inflated bladder
(n=6 in each group). Arrows: Intracellular inclusions. (D) The
inflated bladders were also assessed by SEM at a magnification of
×300 (upper images) and ×900 magnification (middle and lower
images). SEM, scanning electron microscopy. |
Gene expression alterations of mouse
bladder by sodium arsenite
Following 2 weeks of treatment, total RNA was
isolated from bladder tissues derived from control and
arsenic-treated mice by the mouse oligonucleotide DNA microarray
chip. The results indicated 75 upregulated genes and 139
downregulated genes (Table SI). The
data were deposited in the GEO database (accession no. GSE116554).
In order to filter out gene expression related to DNA methylation,
the DNA samples from one control and one arsenic-treated mouse were
analyzed by MeDIP sequencing (Welgene Biotech Co., Ltd.) to obtain
preliminary data (data not shown). After integration analysis of
DNA microarray data, MeDIP sequencing data, PubMed literature
screening, and DNA CpG island searching, 4 cancer-related genes
(Table I) containing potential CpG
island regions (Fig. S2) were
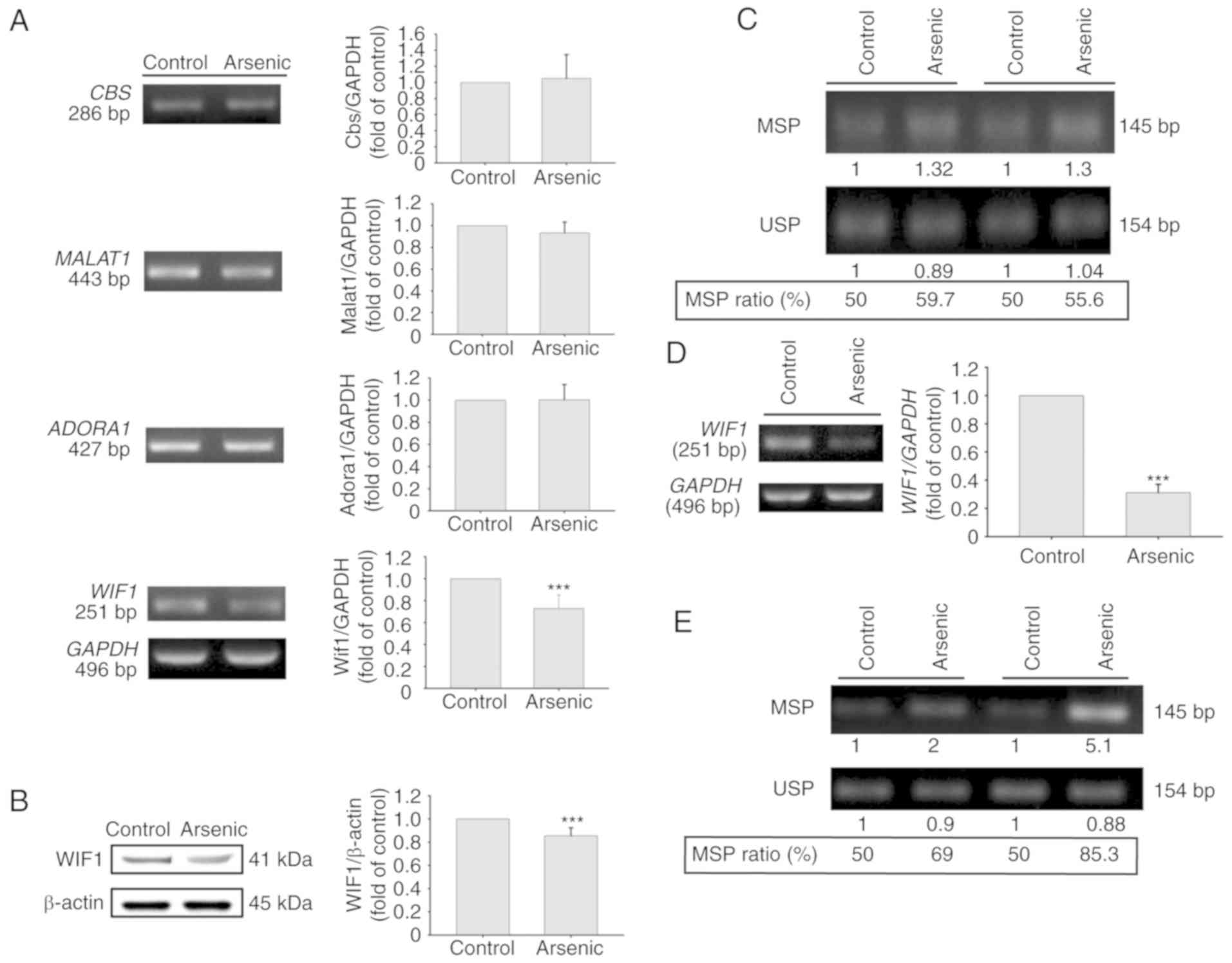
selected for RNA expression verification. CBS, a main metabolic
enzyme which synthesizes H2S, was revealed to be
upregulated in bladder cancer (25),
and its DNA hypomethylation has been revealed to be correlated with
the tumor stage in colorectal cancer (26). MALAT1 is a long noncoding RNA,
which promotes cancer cell proliferation, metastasis and invasion
in several tumor types including bladder cancer and
cholangiocarcinoma (27,28). ADORA1 is a type of adenosine receptor
that is overexpressed in breast cancer (29). An ADORA1 antagonist was reported to
induce MCF-7 cell apoptosis (30).
WIF1 is one of the secreted frizzled-related protein antagonists
that is downregulated in several types of cancer (31). Hypermethylation of WIF1 DNA has
also been reported in bladder cancer (32), chondrosarcoma (33), and non-small cell lung cancer
(34). Quantitative real-time PCR was
applied for RNA detection of these 4 genes including 3 upregulated
(Cbs, Malat1, Adora1) genes and 1 downregulated gene
(Wif1). The results indicated that the mRNA expression
levels of the Cbs and Adora1 genes were significantly
increased by arsenic treatment. The expression levels of
Malat1 were also increased, however the results did not
reveal a significant difference. In contrast to these observations,
the expression levels of Wif1 were slightly decreased, and
no significant difference was obtained between the control and
arsenic-treated mice (Fig. 2A).
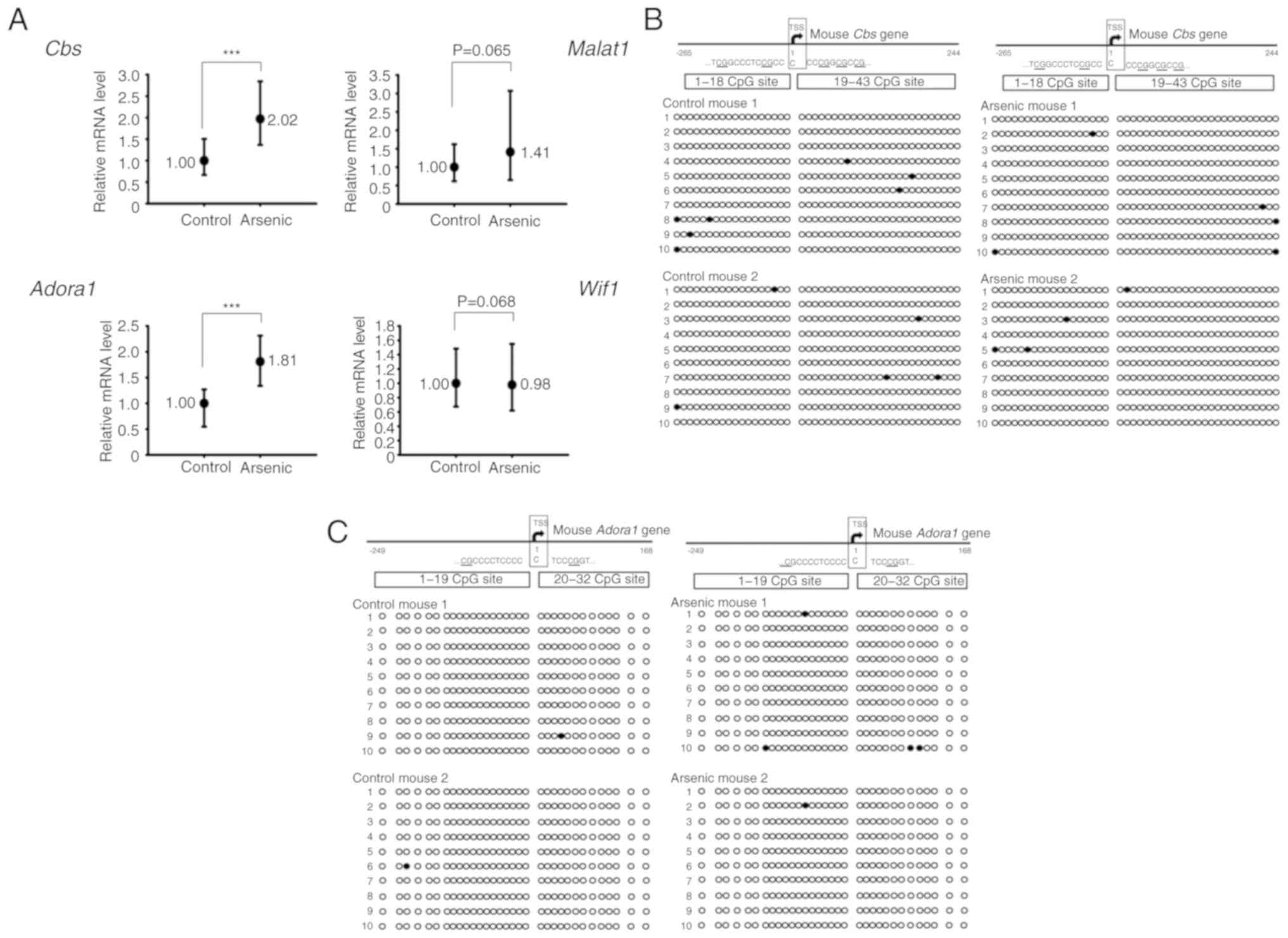 | Figure 2.The relative mRNA expression and DNA
CpG methylation level of selected genes in the mouse bladders. Mice
were administrated 0 ppm (Control, n=6) or 50 ppm arsenic (Arsenic,
n=6) in drinking water for 14 days. (A) After euthanization, the
RNA from the bladders was extracted and analyzed by RT-qPCR.
***P<0.001. (B and C) The DNA CpG methylation pattern of
Cbs and Adora1 genes in the mouse bladders. Mice were
administrated 0 ppm (Control, n=2) or 50 ppm arsenic (Arsenic, n=2)
in drinking water for 14 days. After euthanization, the DNA from
the bladders was extracted and analyzed by BSP assay. Circles
represent a CpG site with methylation (closed circle) or without
methylation (open circle) in the CpG island of (B) Cbs and
(C) Adora1 genes. Cbs, cystathionine β-synthase;
Adora1, adenosine A1 receptor; RT-qPCR, reverse
transcription-quantitative real-time PCR; BSP, bisulfite-sequencing
PCR; Malat1, metastasis-associated lung adenocarcinoma
transcript 1; Wif1, Wnt inhibitory factor 1. |
 | Table I.Normalized intensities of Cbs,
Malat1, Adora1 and Wif1 gene in control and
arsenic-treated mouse bladder from DNA microarray chip
analysis. |
Table I.
Normalized intensities of Cbs,
Malat1, Adora1 and Wif1 gene in control and
arsenic-treated mouse bladder from DNA microarray chip
analysis.
|
| Normalized
Intensity |
|
|
|---|
|
|
|
|
|
|---|
| Gene (ID) | Control | Arsenic | Log2
(arsenic/control) | P-value |
|---|
| Cbs
(12411) |
120 |
252 | 1.147 | 0.00674 |
| Malat1
(72289) | 3982 | 9069 | 1.065 | 0.00208 |
| Adora1
(11539) |
276 |
451 | 0.690 | 0.01485 |
| Wif1
(24117) |
853 |
472 | −0.925 | 0.00079 |
Sodium arsenite did not alter the CpG
island methylation ratio of Cbs and Adora1 genes in mouse bladder
tissues
DNA CpG methylation is one of the main mechanisms
required for gene regulation. The presence of the CpG island near
the transcription start site (TSS) of Cbs and Adora1
genes was used for DNA methylation analysis. The CpG island
methylation levels of Cbs and Adora1 were estimated
to 1.40 and 0.31%, respectively in control mouse bladder tissues.
Following arsenic treatment, the methylation levels noted in
Cbs and Adora1 were 1.05 and 0.78%, respectively
(Fig. 2B and C). The methylation
levels of Cbs and Adora1 were very low (<2%) in
control mice, indicating that these genes were not silenced by CpG
island methylation under normal conditions. Therefore, the gene
activation of Cbs and Adora1 by arsenic may be
mediated by specific transcription factors, translational
activation and/or inhibition of RNA degradation.
Effects of arsenic on gene expression
regulation of CBS, ADORA1, MALAT1 and WIF1 in the human normal
urinary epithelial cell line SV-HUC-1
In addition to the animal studies, the effects of
arsenic on the four selected genes were also analyzed in normal
human urothelial SV-HUC-1 cells. Following arsenic treatment for 14
days, the mRNA expression levels of WIF1 were altered
(Fig. 3A). However, the mRNA
expression levels of ADORA1, CBS and MALAT1 (Fig. 3A) were not altered. Arsenic treatment
decreased WIF1 mRNA and protein expression (Fig. 3A and B). The effects of arsenic on
WIF1 gene methylation were further analyzed in SV-HUC-1
cells. Following 14-day treatment by 0.5 µM of sodium arsenite, the
genomic DNA was extracted for bisulfite conversion and DNA
methylation analysis. Using BSP-MSP/USP analysis, the data
indicated that arsenic slightly increased the MSP ratio (Fig. 3C). This indicated that the decrease in
the levels of WIF1 expression caused by arsenic may be
partly mediated by the increase in the DNA methylation levels. In
order to confirm this hypothesis, the cells were treated with 0.5
µM sodium arsenite for 10 weeks. The results indicated that
WIF1 mRNA levels were further decreased (Fig. 3D) and that the MSP ratio was
considerably increased (Fig. 3E). The
inverse correlation between MSP ratio and WIF1 mRNA
expression was also reported in bladder tumor samples from primary
transitional cell carcinoma patients (32).
Effects of arsenic on cellular
functions in the human normal urinary epithelial cell line
SV-HUC-1
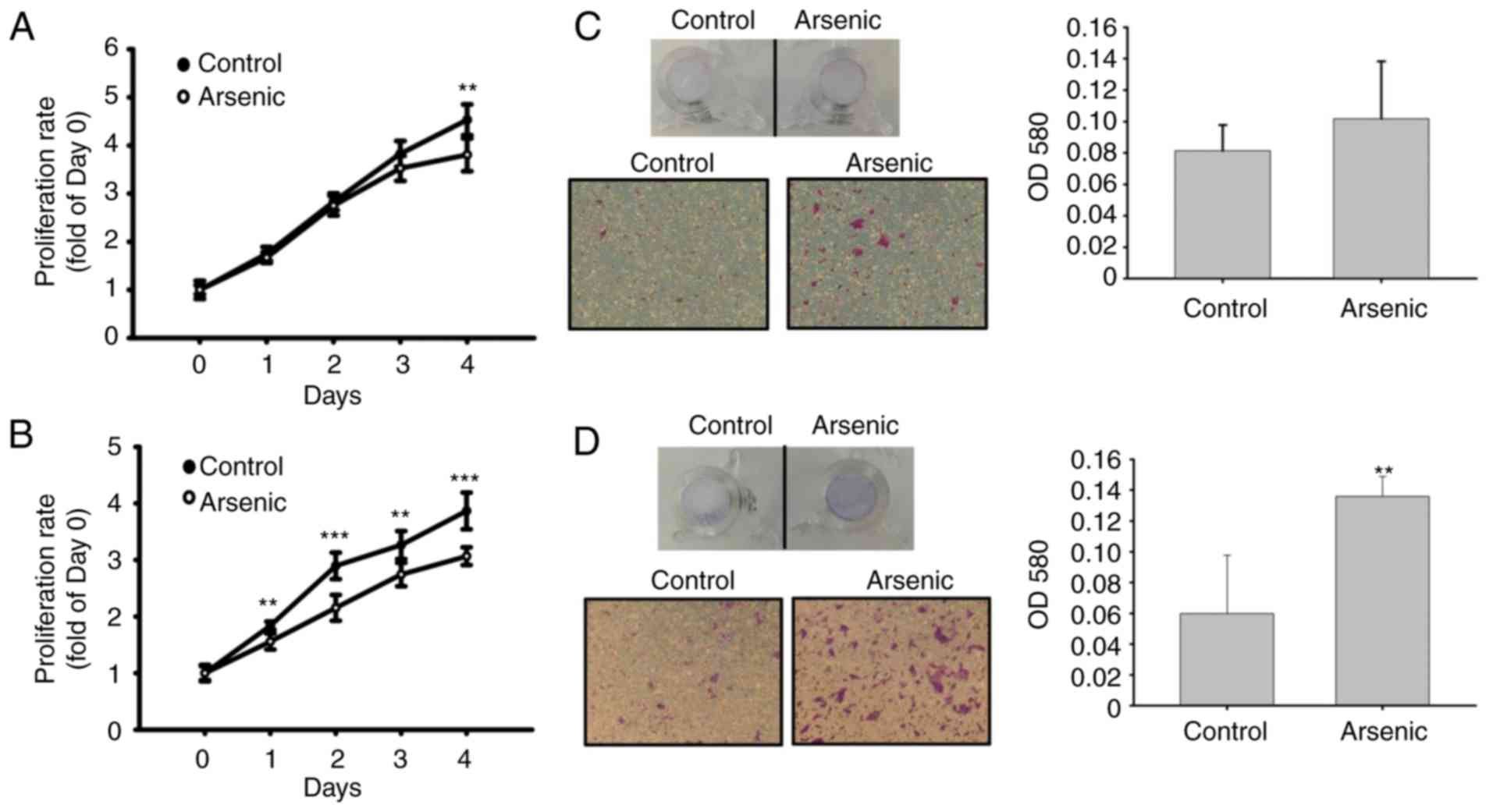
Cell proliferative activity was analyzed to assess
arsenic-induced urothelial hyperplasia in mice. Arsenic caused a
decrease in cell proliferation (Fig. 4A
and B). The migratory activity was further analyzed by
Transwell assay. Following a 14-day and 10-week treatment with
arsenic of the cells, the migratory ability of the urothelial cells
was slightly (14-day, Fig. 4C) and
significantly (10-week, Fig. 4D)
increased. This indicated that sodium arsenite promoted urothelial
migration following long-term treatment, although it was capable of
inhibiting cell proliferation.
Gene expression profile and DNA CpG
methylation analysis of the WIF1 gene in various human urinary
epithelial cell lines
The mRNA expression and the DNA methylation levels
of WIF1 were assayed in various human urothelial cell lines
in order to examine the differential expression of this gene in
normal and cancer tissues. The 7 urothelial carcinoma cell lines,
namely 5637, J82, RT4, 1376, T24, TSGH8301 and BFTC905 expressed
lower levels of WIF1 mRNA (Fig.
5A) compared with those observed in the normal urothelial cell
line SV-HUC-1. In addition, these cell lines expressed higher
levels of DNA MSP ratio, with the exception of 5637, compared with
the corresponding normal cell line (Fig.
5B). The DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine
increased WIF1 expression in 5 bladder cancer cell lines
(Fig. 5C), indicating that
WIF1 gene expression was inhibited in urothelial carcinoma
cells, possibly as a result of DNA CpG methylation.
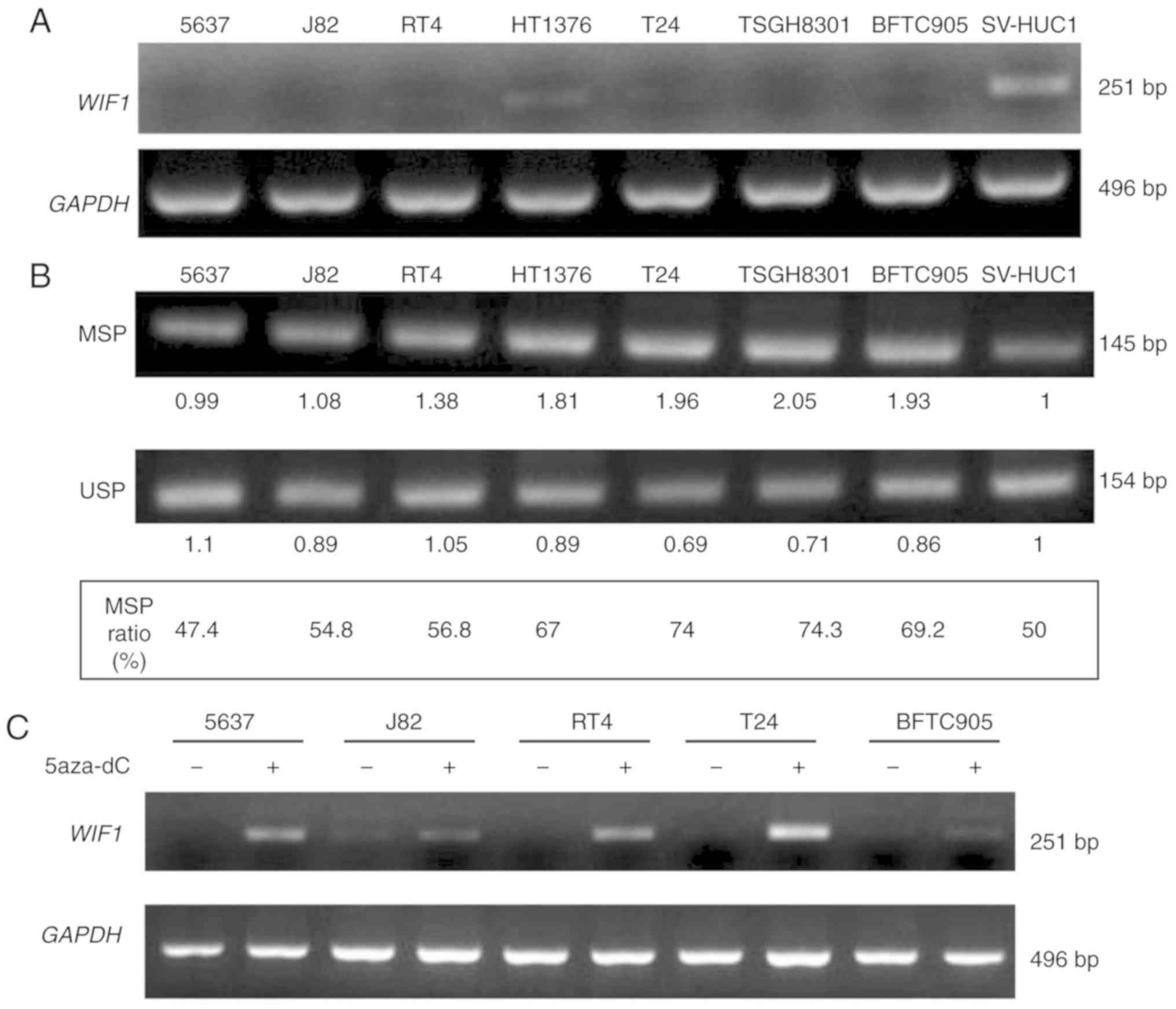 | Figure 5.The RNA expression level and DNA CpG
methylation level of WIF1 in 8 human urothelial cell lines.
(A) The mRNA expression level of WIF1 was analyzed by RT-PCR
assay. (B) The DNA methylation level of WIF1 was analyzed by
BSP-MSP/USP assay. The MSP ratio (%)=MSP intensity/MSP intensity +
USP intensity. Seven human bladder cancer cell lines (5637, J82,
RT4, 1376, T24, TSGH8301, BFTC905) and one normal human urothelial
cell line (SV-HUC1) were analyzed concurrently. (C) WIF1
mRNA expression after 5-aza-2′-deoxycytidine treatment for 3 days.
WIF1, Wnt inhibitory factor 1; BSP, bisulfite-sequencing
PCR; MSP, methylation-specific PCR; USP, unmethylation-specific
PCR. |
Discussion
In the present study, it was demonstrated that
arsenic increased the expression levels of mouse Cbs and
Adora1 regardless of the levels of DNA methylation, while it
concomitantly decreased human WIF1 expression partly by
increasing DNA CpG methylation. The present data further suggested
that the regulation of arsenic-induced gene expression was not the
same between different species (mice vs. humans), which may have
accounted for the differences noted in vivo and in
vitro. It has been revealed that inorganic arsenic is
metabolized to organic arsenic in specific organisms by multiple
pathways including glutathione and methyl conjugation (35). However, these effects have not been
revealed in cells derived from human bladder tissues (36). The data reported in the present study
indicated that 50 ppm arsenic (666 µM sodium arsenite) did not
affect mouse body weight, although 0.5 µM of sodium arsenite
retarded cell proliferation in SV-HUC-1 cells. Therefore, in
vivo, sodium arsenite may be metabolized to organic arsenic,
which is less cytotoxic than the inorganic arsenic form noted in
cultured cells (37). In our
experience, the toxicity of 0.5 µM sodium arsenite is similar to
100 µM dimethyl arsenic acid in cultured bladder cells. Therefore,
not only cytotoxicity, but also the biological response of arsenic
metabolities (e.g. dimethyl arsenic) should be different from
sodium arsenite.
Oral arsenic but not local treatment relates to a
higher odds ratio of developing bladder, lung and liver cancers in
human (9), and induces carcinogenesis
of bladder, liver (37) and lung
(38) in animal models. In the
present study, short-time arsenic exposure only induced
histological change but not tumor formation in the bladder and
liver. In theory, there are multiple enzymes in liver cells for
drug metabolism including inorganic arsenic, and liver damage may
affect arsenic metabolism. Different arsenic metabolites may alter
the response of arsenic in the bladder. Therefore, arsenic-induced
liver damage may affect the toxicity of arsenic to the bladder. As
for the lung, a limitation of the present study was that there was
no histological examination on lung tissue.
KEGG Pathway analysis of this mouse gene expression
data resulted 12 geneset pathways (Table
SII). Although WNT signaling was not revealed in the results of
the pathway analysis; however, Gene Ontology analysis concluded
that WIF1 belongs to negative regulation of the WNT signaling
pathway (GO:0030178). WIF1 is one of the functional inhibitors of
the WNT pathway (39). The WNT
signaling pathway is generally divided into 3 subpathways as
follows: Canonical, non-canonical planar cell polarity (PCP), and
non-canonical WNT/calcium (40). In
the WNT-activated canonical pathway, β-catenin accumulates in the
cytoplasm and translocates to the nucleus in order to cause
activation of specific genes including the epithelial-mesenchymal
transition (EMT)-related genes. The PCP pathway affects the
cytoskeleton and triggers the expression of target genes
responsible for cell adhesion and migration. In the
calcium-dependent pathway, WNT further enhances cell migration by
the activation of calmodulin. The WNT signaling complex originates
from multiple membrane receptors, which are extracellularly
regulated by various secreted antagonists, including Cerberus,
Dickopf-related protein 1 (DKK1), secreted Frizzled-related protein
(SFRP) and Sclerostin/Wise and Wnt inhibitory factor (WIF)
(41). Among these WNT antagonists,
SFRP (42) and WIF1
(32) have been reported to possess
lower mRNA expression levels and higher promoter methylation levels
in bladder tumor samples than those noted in the corresponding
normal tissues. In addition to bladder cancer, WIF1 DNA
hypermethylation has been identified as a potential biomarker for
the diagnosis of lung cancer (32)
and chondrosarcoma (33). Since WIF1
inhibits WNT-related cell migration, reduced WIF1 expression may be
one of the mechanisms involved in arsenic-induced cell migration
(Fig. 4C and D). By performing a
search in the Oncomine database, it was demonstrated that the mRNA
expression levels of WIF1 exhibited a significant decrease
in the infiltration of bladder urothelial carcinoma compared with
that noted in superficial bladder cancer (Fig. S3). The data indicated that
WIF1 downregulation was associated with bladder cancer
progression.
CBS is the enzyme catalyzing the first step of the
conversion of homocysteine to cysteine. In addition, it further
catalyzes the metabolism of homocysteine and cysteine to
cystathionine and hydrogen sulfide (43). In the present study, arsenic increased
Cbs mRNA expression without altering its DNA methylation
levels (Fig. 2A and B). The DNA
methylation levels of Cbs were low in normal mouse bladder
tissues (Fig. 2B), possibly due to
the gene expression not being affected by DNA demethylation. ADORA1
is a membrane receptor expressed ubiquitously in the body. Similar
to Cbs, Adora1 DNA methylation levels were also low in
normal mouse bladder tissues (Fig.
2C). Therefore, the increase in the levels of Adora1 was
not mediated by DNA demethylation. The contribution of
ADORA1 in cancer progression remains controversial. For
example, the application of ADORA1 antagonists in MCF-7 breast
cancer cells induced cell apoptosis via the induction of p53
expression (30); however, in
colorectal cancer, metformin induced apoptosis via the
ADORA1/AMPK/mTOR pathway (44). In
the present study, the expression levels of arsenic-induced
Cbs and Adora1 genes were upregulated in mouse
bladder tissues and not in human urothelial cells. Using Oncomine
search, the absence of differential expression in CBS and
ADORA1 mRNA levels in bladder cancer tissues [bladder cancer
database, threshold by: P-value (1E-4), fold change (1.5) and gene
rank (all)] was demonstrated.
Animal experiments are an essential step prior to
the conduct of clinical studies. The main species used in animal
studies is rodents. Despite the extensive use of rodents in
research, in some cases concerns are raised regarding the
discrepancies between animal models and human population studies.
For example, in the case of arsenic, it is not easy to induce
cancer in rodents unless a high dosage of this compound is used
(12). In contrast to these
observations, several epidemiological studies have suggested that
arsenic is a pivotal factor for the development of human cancer
(45,46). Our previous study demonstrated that
N-butyl-N-(4-hydroxybutyl) nitrosamine (BBN) caused a
decrease in glutathione S-transferase Mu1 (Gstm1) levels by
downregulating the expression of the corresponding gene (47). However, the GSTM1 gene is
deleted in approximately 50% of humans (48), whereas this percentage is not found in
mice. The gene regulation of GSTM1 is different between mice
and humans. Analysis of the gene expression changes of BBN-induced
bladder cancer in mice and rats revealed that approximately 6% of
genes shared the same regulation pattern in these two species
(49). This indicated that the
regulation of carcinogen-induced bladder cancer genes was distinct
in mice and rats. Therefore, it may be assumed that these
differences are also present between rodents and humans. In a
similar study, it was revealed that 25 mg/kg of ketamine induced
rat bladder inflammation and cyclooxygenase-2 expression (50), whereas 100 mg/kg of ketamine did not
cause significant inflammation in mouse bladder tissues (51). In the present study, we provide
additional evidence that the gene regulation of CBS, ADORA1
and WIF1 was different between mouse bladder tissues and
human urothelial cells. Therefore, the extrapolation of rodent to
human data should be assessed with caution.
In summary (Fig. 6),
the present study demonstrated that sodium arsenite induced gene
expression alteration, hyperplasia and dysplasia in mouse
urothelium. Arsenite increased Cbs and Adora1 gene
expression without affecting DNA methylation levels in the mouse
bladder. In contrast to these two genes, sodium arsenite reduced
WIF1 gene expression in human urothelium, and increased its
DNA methylation levels, which may play a role in the gene
downregulation. In addition, the downregulation of the WIF1
gene may increase cell migration. Finally, the decrease in gene
expression and the increase in DNA methylation of WIF1 could
be considered potential biomarkers for the diagnosis of human
bladder cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Ministry of Science and Technology of the Republic of China, Taiwan
MOST 107-2320-B-415-001 and from Ditmanson Medical Foundation
Chiayi Christian Hospital Research Program R107-002, Taiwan.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author upon
reasonable request.
Authors' contributions
YCJ, SCW, and YWL were involved in the design of
the study, acquisition, and interpretation of data and drafting the
article. SCW and SYC performed the experiments. YCD and CHS
assisted histopathologic analysis. YCJ and YWL analyzed and
discussed the data. YRL and LCC assisted in the statistical
analysis and interpretation of the data and revised the article.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All animal experiments were performed according to
the national and institutional guidelines and were approved by the
Institutional Animal Care and Use Committee of National Chiayi
University (approval no. 103040.1) authorized by the local
government for the regulation of animal welfare.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ADORA1
|
adenosine A1 receptor
|
|
BSP
|
bisulfite-sequencing PCR
|
|
CBS
|
cystathionine β-synthase
|
|
DEGs
|
differentially expressed genes
|
|
DMA
|
dimethylarsinous acid
|
|
DMSO
|
dimethyl sulfoxide
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
GEO
|
Gene Expression Omnibus
|
|
IARC
|
International Agency for Research on
Cancer
|
|
MALAT1
|
metastasis-associated lung
adenocarcinoma transcript 1
|
|
MMA
|
monomethylarsonous acid
|
|
MSP
|
methylation-specific PCR
|
|
MTT
|
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
SEM
|
scanning electron microscopy
|
|
USEPA
|
United States Environmental
Protection Agency
|
|
USP
|
unmethylation-specific PCR
|
|
WIF1
|
Wnt inhibitory factor 1
|
References
|
1
|
Hughes MF: Arsenic toxicity and potential
mechanisms of action. Toxicol Lett. 133:1–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
IARC, . Arsenic and inorganic arsenic
compounds. http://monographs.iarc.fr/ENG/Classification/latest_classif.phpWorld
Health Organization. 1-121:41–94. 2012.
|
|
3
|
Lee-Feldstein A: Cumulative exposure to
arsenic and its relationship to respiratory cancer among copper
smelter employees. J Occup Med. 28:296–302. 1986.PubMed/NCBI
|
|
4
|
Tseng WP: Effects and dose-response
relationships of skin cancer and blackfoot disease with arsenic.
Environ Health Perspect. 19:109–119. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cebrian ME, Albores A, Aguilar M and
Blakely E: Chronic arsenic poisoning in the north of Mexico. Hum
Toxicol. 2:121–133. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chiou HY, Chiou ST, Hsu YH, Chou YL, Tseng
CH, Wei ML and Chen CJ: Incidence of transitional cell carcinoma
and arsenic in drinking water: A follow-up study of 8,102 residents
in an arseniasis-endemic area in northeastern Taiwan. Am J
Epidemiol. 153:411–418. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hopenhayn-Rich C, Biggs ML, Fuchs A,
Bergoglio R, Tello EE, Nicolli H and Smith AH: Bladder cancer
mortality associated with arsenic in drinking water in Argentina.
Epidemiology. 7:117–124. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith AH, Goycolea M, Haque R and Biggs
ML: Marked increase in bladder and lung cancer mortality in a
region of Northern Chile due to arsenic in drinking water. Am J
Epidemiol. 147:660–669. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen CJ, Chuang YC, You SL, Lin TM and Wu
HY: A retrospective study on malignant neoplasms of bladder, lung
and liver in blackfoot disease endemic area in Taiwan. Br J Cancer.
53:399–405. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tokar EJ, Benbrahim-Tallaa L, Ward JM,
Lunn R, Sams RL II and Waalkes MP: Cancer in experimental animals
exposed to arsenic and arsenic compounds. Crit Rev Toxicol.
40:912–927. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei M, Wanibuchi H, Morimura K, Iwai S,
Yoshida K, Endo G, Nakae D and Fukushima S: Carcinogenicity of
dimethylarsinic acid in male F344 rats and genetic alterations in
induced urinary bladder tumors. Carcinogenesis. 23:1387–1397. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cohen SM, Ohnishi T, Arnold LL and Le XC:
Arsenic-induced bladder cancer in an animal model. Toxicol Appl
Pharmacol. 222:258–263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arnold LL, Eldan M, Nyska A, van Gemert M
and Cohen SM: Dimethylarsinic acid: Results of chronic
toxicity/oncogenicity studies in F344 rats and in B6C3F1 mice.
Toxicology. 223:82–100. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yokohira M, Arnold LL, Pennington KL,
Suzuki S, Kakiuchi-Kiyota S, Herbin-Davis K, Thomas DJ and Cohen
SM: Effect of sodium arsenite dose administered in the drinking
water on the urinary bladder epithelium of female arsenic (+3
oxidation state) methyltransferase knockout mice. Toxicol Sci.
121:257–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arnold LL, Suzuki S, Yokohira M,
Kakiuchi-Kiyota S, Pennington KL and Cohen SM: Time course of
urothelial changes in rats and mice orally administered arsenite.
Toxicol Pathol. 42:855–862. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dai YC, Wang SC, Haque MM, Lin WH, Lin LC,
Chen CH and Liu YW: The interaction of arsenic and
N-butyl-N-(4-hydroxybutyl)nitrosamine on urothelial carcinogenesis
in mice. PLoS One. 12:e01862142017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su PF, Hu YJ, Ho IC, Cheng YM and Lee TC:
Distinct gene expression profiles in immortalized human urothelial
cells exposed to inorganic arsenite and its methylated trivalent
metabolites. Environ Health Perspect. 114:394–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Medeiros M, Zheng X, Novak P, Wnek SM,
Chyan V, Escudero-Lourdes C and Gandolfi AJ: Global gene expression
changes in human urothelial cells exposed to low-level
monomethylarsonous acid. Toxicology. 291:102–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boivin GP, Bottomley MA, Schimi PA, Goss L
and Grobe N: Physiologic, behavioral, and histologic responses to
various euthanasia methods in C57BL/6NTac male mice. J Am Assoc Lab
Anim Sci. 56:69–78. 2017.PubMed/NCBI
|
|
20
|
Leary S, Underwood W, Anthony R, Cartner
S, Corey D, Grandin T, Greenacre C, Gwaltney-Brant S, MaCrackin MA,
Meyer R, et al: AVMA guidelines for the euthanasia of animals.
2013.edition.
|
|
21
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG; National centre for the replacement refinement and
reduction of animals in research, : Animal research: Reporting in
vivo experiments-the ARRIVE guidelines. J Cereb Blood Flow Metab.
31:991–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weng L, Dai H, Zhan Y, He Y, Stepaniants
SB and Bassett DE: Rosetta error model for gene expression
analysis. Bioinformatics. 22:1111–1121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen CH, Wang ST, Wang SC, Lin SM, Lin LC,
Dia YC and Liu YW: Ketamine-induced bladder dysfunction is
associated with extracellular matrix accumulation and impairment of
clacium signaling in a mouse model. Mol Med Rep. 19:2716–2728.
2019.PubMed/NCBI
|
|
24
|
Dodmane PR, Arnold LL, Muirhead DE, Suzuki
S, Yokohira M, Pennington KL, Dave BJ, Lu X, Le XC and Cohen SM:
Characterization of intracellular inclusions in the urothelium of
mice exposed to inorganic arsenic. Toxicol Sci. 137:36–46. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gai JW, Qin W, Liu M, Wang HF, Zhang M, Li
M, Zhou WH, Ma QT, Liu GM, Song WH, et al: Expression profile of
hydrogen sulfide and its synthases correlates with tumor stage and
grade in urothelial cell carcinoma of bladder. Urol Oncol.
34:166.e15–e20. 2016. View Article : Google Scholar
|
|
26
|
Xue G, Lu CJ, Pan SJ, Zhang YL, Miao H,
Shan S, Zhu XT and Zhang Y: DNA hypomethylation of CBS promoter
induced by folate deficiency is a potential noninvasive circulating
biomarker for colorectal adenocarcinomas. Oncotarget.
8:51387–51401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li C, Cui Y, Liu LF, Ren WB, Li QQ, Zhou
X, Li YL, Li Y, Bai XY and Zu XB: High expression of long noncoding
RNA MALAT1 indicates a poor prognosis and promotes clinical
progression and metastasis in bladder cancer. Clin Genitourin
Cancer. 15:570–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang C, Mao ZP, Wang L, Wu GH, Zhang FH,
Wang DY and Shi JL: Long non-coding RNA MALAT1 promotes
cholangiocarcinoma cell proliferation and invasion by activating
PI3K/Akt pathway. Neoplasma. 64:725–731. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mirza A, Basso A, Black S, Malkowski M,
Kwee L, Pachter JA, Lachowicz JE, Wang Y and Liu S: RNA
interference targeting of A1 receptor-overexpressing breast
carcinoma cells leads to diminished rates of cell proliferation and
induction of apoptosis. Cancer Biol Ther. 4:1355–1360. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dastjerdi MN, Rarani MZ, Valiani A and
Mahmoudieh M: The effect of adenosine A1 receptor agonist and
antagonist on p53 and caspase 3, 8, and 9 expression and apoptosis
rate in MCF-7 breast cancer cell line. Res Pharm Sci. 11:303–310.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wissmann C, Wild PJ, Kaiser S, Roepcke S,
Stoehr R, Woenckhaus M, Kristiansen G, Hsieh JC, Hofstaedter F,
Hartmann A, et al: WIF1, a component of the Wnt pathway, is
down-regulated in prostate, breast, lung, and bladder cancer. J
Pathol. 201:204–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Urakami S, Shiina H, Enokida H, Kawakami
T, Tokizane T, Ogishima T, Tanaka Y, Li LC, Ribeiro-Filho LA,
Terashima M, et al: Epigenetic inactivation of Wnt inhibitory
factor-1 plays an important role in bladder cancer through aberrant
canonical Wnt/beta-catenin signaling pathway. Clin Cancer Res.
12:383–391. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu P, Shen JK, Hornicek FJ, Liu F and
Duan Z: Wnt inhibitory factor 1 (WIF1) methylation and its
association with clinical prognosis in patients with
chondrosarcoma. Sci Rep. 7:15802017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo H, Zhou S, Tan L, Wu X, Wu Z and Ran
R: Clinicopathological significance of WIF1 hypermethylation in
NSCLC, a meta-analysis and literature review. Oncotarget.
8:2550–2557. 2017.PubMed/NCBI
|
|
35
|
Watanabe T and Hirano S: Metabolism of
arsenic and its toxicological relevance. Arch Toxicol. 87:969–979.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Styblo M, Del Razo LM, Vega L, Germolec
DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR and Thomas
DJ: Comparative toxicity of trivalent and pentavalent inorganic and
methylated arsenicals in rat and human cells. Arch Toxicol.
74:289–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wanibuchi H, Salim EI, Kinoshita A, Shen
J, Wei M, Morimura K, Yoshida K, Kuroda K, Endo G and Fukushima S:
Understanding arsenic carcinogenicity by the use of animal models.
Toxicol Appl Pharmacol. 198:366–376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cui X, Wakai T, Shirai Y, Hatakeyama K and
Hirano S: Chronic oral exposure to inorganic arsenate interferes
with methylation status of p16INK4a and RASSF1A and induces lung
cancer in A/J mice. Toxicol Sci. 91:372–381. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawano Y and Kypta R: Secreted antagonists
of the Wnt signalling pathway. J Cell Sci. 116:2627–2634. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Duchartre Y, Kim YM and Kahn M: The Wnt
signaling pathway in cancer. Crit Rev Oncol Hematol. 99:141–149.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Niehrs C: The complex world of WNT
receptor signalling. Nat Rev Mol Cell Biol. 13:767–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang X, Wang H, Bu R, Fei X, Zhao C and
Song Y: Methylation and aberrant expression of the Wnt antagonist
secreted Frizzled-related protein 1 in bladder cancer. Oncol Lett.
4:334–338. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jhee KH and Kruger WD: The role of
cystathionine beta-synthase in homocysteine metabolism. Antioxid
Redox Signal. 7:813–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lan B, Zhang J, Zhang P, Zhang W, Yang S,
Lu D, Li W and Dai Q: Metformin suppresses CRC growth by inducing
apoptosis via ADORA1. Front Biosci (Landmark Ed). 22:248–257. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Baris D, Waddell R, Beane Freeman LE,
Schwenn M, Colt JS, Ayotte JD, Ward MH, Nuckols J, Schned A,
Jackson B, et al: Elevated bladder cancer in northern New England:
The role of drinking water and arsenic. J Natl Cancer Inst.
108(pii): djw0992016.PubMed/NCBI
|
|
46
|
Saint-Jacques N, Parker L, Brown P and
Dummer TJ: Arsenic in drinking water and urinary tract cancers: A
systematic review of 30 years of epidemiological evidence. Environ
Health. 13:442014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chuang JJ, Dai YC, Lin YL, Chen YY, Lin
WH, Chan HL and Liu YW: Downregulation of glutathione S-transferase
M1 protein in N-butyl-N-(4-hydroxybutyl)nitrosamine-induced mouse
bladder carcinogenesis. Toxicol Appl Pharmacol. 279:322–330. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Engel LS, Taioli E, Pfeiffer R,
Garcia-Closas M, Marcus PM, Lan Q, Boffetta P, Vineis P, Autrup H,
Bell DA, et al: Pooled analysis and meta-analysis of glutathione
S-transferase M1 and bladder cancer: A HuGE review. Am J Epidemiol.
156:95–109. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Williams PD, Lee JK and Theodorescu D:
Molecular credentialing of rodent bladder carcinogenesis models.
Neoplasia. 10:838–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Juan YS, Lee YL, Long CY, Wong JH, Jang
MY, Lu JH, Wu WJ, Huang YS, Chang WC and Chuang SM: Translocation
of NF-κB and expression of cyclooxygenase-2 are enhanced by
ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol.
185:2269–2285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shen CH, Wang SC, Wang ST, Lin SM, Wu JD,
Lin CT and Liu YW: Evaluation of urinary bladder fibrogenesis in a
mouse model of long-term ketamine injection. Mol Med Rep.
14:1880–1890. 2016. View Article : Google Scholar : PubMed/NCBI
|