Introduction
Bladder cancer (BC) is the most frequent neoplasm of
the urinary tract (1). At
diagnosis, ~75% of cases are non-muscle-invasive BC, while 25% of
cases present with muscle-invasive BC (2). The first-line treatment is
cisplatin-containing combination chemotherapy such as gemcitabine
plus cisplatin or methotrexate, vinblastine, doxorubicin and
cisplatin (2,3). Paclitaxel (PTX) has been recently
reported to be effective in inhibiting BC (4–6).
Clinical trials have revealed that PTX combined with radiation
(7) or gemcitabine (8) are effective treatment strategies for
patients with BC, indicating that PTX is a promising second-line
treatment option for patients with metastatic BC. However, a
significant proportion of patients will relapse due to development
of drug resistance to the chemotherapeutic regimens (9,10).
Cellular senescence is irreversible cell cycle
arrest in response to various forms of cellular stresses (11). In contrast to the well-studied
replicative senescence of somatic cells, therapeutic implications
and mechanisms of senescence in cancer treatment remain elusive
(12). Currently, it is generally
accepted that senescence is a tumor-suppressive mechanism, which
restricts the unlimited cell proliferation, thus preventing the
occurrence and development of cancer (13). Cancer cells may undergo senescence
in response to ionizing radiation or chemotherapy, known as
therapy-induced senescence (TIS) (14). Moreover, TIS may act as a ‘back-up’
response to cancer therapy, in which apoptotic pathways are
disabled (14,15). PTX has been reported to induce
senescence of breast cancer cells (16,17).
However, to the best of our knowledge, TIS has not yet been
reported in BC.
The circadian clock is an intrinsic timekeeping
system that regulates multiple vital physiological and biochemical
processes, including cell proliferation and senescence (18,19).
The core clock genes include circadian locomotor output cycles
kaput (CLOCK), brain and muscle Arnt-like protein 1 (BMAL1), period
(PER)1/2 and cryptochrome (CRY)1/2, which constitute a
transcriptional auto-regulatory feedback loop (20). Disruption of the circadian clock can
increase cancer risk in humans, but the effect of each of the four
core circadian genes on tumor is not always consistent in tumors
from different human organs (21–23).
Furthermore, the relationship between the circadian clock and drug
resistance is yet to be fully understood. The present study aimed
to investigate the circadian clock in cisplatin-resistant (Res) BC
cells and to examine the regulatory effect of clock genes on
PTX-induced senescence.
Materials and methods
Cell culture and drug treatment
Human BC UMUC3 and EJ cell lines were purchased from
the American Type Culture Collection (ATCC). EJ cells were
authenticated by high-resolution small tandem repeat profiling and
were confirmed mycoplasma-free before experiments began. Cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
penicillin (100 U/ml) and streptomycin (100 µg/ml), at 37°C in a
balanced air humidified incubator with an atmosphere of 5%
CO2. Res cell lines were established via long term
incubation (2 months) at 37°C with increasing concentration of
cisplatin (Sigma-Aldrich; Merck KGaA) in a range of 0–4 µg/ml, and
then were steadily grown in the presence of 2 µg/ml cisplatin. For
PTX treatment, the Res cells or the parental UMUC3 and EJ cells
were treated with 80 or 40 nM PTX (Sigma-Aldrich; Merck KGaA),
respectively, for 3 days. For serum-starvation, cells were treated
with serum-free medium for 48 h and then synchronized with 0.1 µM
dexamethasone for 2 h to examine the circadian rhythm. Cells were
incubated with serum-free medium for 24, 48 and 72 h, and the
circadian proteins were examined via western blotting.
Cell synchronization
Cells were synchronized in circadian time via
aspiration of media and replacement with fresh DMEM containing 0.1
µM dexamethasone (Sigma-Aldrich; Merck KGaA). The cells were
synchronized for 2 h at 37°C, followed by replacing with fresh
medium (circadian time 0; ZT0). The cells did not receive any
further medium changes from this point until the time of harvest.
Individual plates were harvested for total RNA at ZT0, ZT4, ZT8,
ZT12, ZT16, ZT20, ZT24, ZT28, ZT32, ZT36, ZT40, ZT44 and ZT48.
Statistical analysis was cosine fitted using OriginPro 8.0
(https://www.originlab.com/).
Flow cytometry
For cell cycle analysis, cells were fixed in 70%
ethanol overnight at 4°C and then were washed twice with ice-cold
PBS. Cells were stained with PI staining solution (50 µg/ml PI, 100
µg/ml RNase, 0.05% Triton X-100 in ddH2O) at room temperature for
20–30 min. PI-stained cells were analyzed for their DNA content
using FACSCalibur flow cytometer (BD Biosciences) and the data
analyzed using FlowJo 7.6 (www.flowjo.com).
Cellular apoptosis was measured using the Annexin
V-FITC Apoptosis Detection kit (Invitrogen; Thermo Fisher
Scientific, Inc.). Briefly, 1×105 cells were resuspended
in Annexin V binding buffer and stained with Annexin V-FITC and PI
(1 µg/ml) at room temperature for 15 min. The apoptotic rate was
quantified via FACSCalibur flow cytometer (BD Biosciences) and the
data analyzed using FlowJo 7.6 (www.flowjo.com).
MTS assay
To assess cell viability, the cells were seeded
(5×103 cells/well) into 96-well plates and incubated at
37°C with 5% CO2 for 24 h. Then, the cells were treated
with increasing concentration of cisplatin (0–40 µg/ml) or PTX
(0-1,000 nM) for 48 h at 37°C. After incubation, 20 µl MTS solution
was added into each well and incubated for 2 h at 37°C. The number
of viable cells was evaluated by measuring the absorbance at 490 nm
with a microplate reader (Thermo Fisher Scientific, Inc.). In total
≥3 independent experiments were conducted.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells with
TRIzol® reagent (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. cDNA was synthesized with
PrimeScript™ RT Master mix (Takara Biotechnology Co., Ltd.) for 15
min at 37°C, and then the reverse transcriptase was inactivated for
5 sec at 85°C. qPCR was performed with SYBR® Premix Ex
Taq™ II (Tli RNaseH Plus; Takara Biotechnology Co., Ltd.) with the
following conditions in an Applied Biosystems instrument: Initial
denaturation at 95°C for 30 sec, followed by 40 cycles of 95°C for
10 sec, 60°C for 5 sec and 72°C for 15 sec, 95°C for 15 sec, 60°C
for 60 sec. The samples were quantified with 2−ΔΔCq
method and β-actin was used as internal reference (24). The following primers were used:
BMAL1 forward, 5′-TGCAACGCAATGTCCAGGAA-3′ and reverse,
5′-GGTGGCACCTCTTAATGTTTTCA-3′; CLOCK forward,
5′-TGCGAGGAACAATAGACCCAA−3′ and reverse,
5′-ATGGCCTATGTGTGCGTTGTA−3′; CRY1 forward,
5′-TTGGAAAGGAACGAGACGCAG−3′ and reverse,
5′-CGGTTGTCCACCATTGAGTT-3′; PER2 forward,
5′-GACATGAGACCAACGAAAACTGC-3′ and reverse,
5′-AGGCTAAAGGTATCTGGACTCTG-3′; β-Actin forward,
5′-CATGTACGTTGCTATCCAGGC−3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT−3′; and p53 forward,
5′-CAGCACATGACGGAGGTTGT-3′ and reverse
5′-TCATCCAAATACTCCACACGC−3′.
Western blotting
Total proteins from cells were extracted with RIPA
lysis buffer containing 1 mM PMSF (Fude: http://www.fdbio.net) and phosphatase inhibitor
(Fude). Protein concentration was measured using a BCA Protein
Assay kit (CWBio: http://www.cwbiotech.bioon.com.cn). The proteins (50
µg/lane) were loaded on 10% SDS-PAGE and separated by
electrophoresis, followed by blotting on a PVDF membrane (EMD
Millipore). The membrane was blocked in Tris-buffered saline (pH
8.0)+0.1% Tween-20 and 5% skim milk at room temperature for 2 h.
The corresponding primary antibody (1:1,000) was incubated
overnight at 4°C and then incubated with the horseradish
peroxide-conjugated secondary antibody (Fude; 1:10,000) at room
temperature for 1 h. Immunological signals were detected using an
electrochemical luminescence kit (Yeasen; www.yeasen.com). The band intensities were quantified
using Image-Pro-Plus 6.0 software (MediaCybernetics, Inc.). The
primary antibodies are as follows: CRY1 (cat. no. ab245564; Abcam),
PER2 (cat. no. ab179813; Abcam), CLOCK (cat. no. 18094-1-AP;
ProteinTech Group, Inc.), BMAL1 (cat. no. 14268-1-AP; ProteinTech
Group, Inc.), p53 (cat. no. 2524; Cell Signaling Technology, Inc.),
p21 (cat. no. 10355-1-AP; ProteinTech Group, Inc.) and GAPDH (cat.
no. AP0063; Biogot Technology Co., Ltd.).
5-Ethynyl-2′-deoxyuridine (EdU)
incorporation assays
Cell proliferation was detected with a EdU
labeling/detection kit (Guangzhou RiboBio Co., Ltd.). Cells were
plated in 24-well plates (1×105 cells/well). EdU
labeling medium (1:1,000) was added into wells and incubated for 2
h at 37°C according to the manufacturer's instructions. After EdU
staining, cells were counterstained with Hoechst 33342 for 30 min
at room temperature. The percentage of EdU+ cells was
calculated from five random fields each in three wells via
fluorescence microscopy (magnification, ×400; Nikon
Corporation).
Small interfering RNA (siRNA)
transfection
Cry1-targeting siRNAs (si-Cry1-1,
5′-GATGCAGATTGGAGCATAA-3′; and si-Cry1-2,
5′-GGATGAAACAGATCTATCA-3′) were designed by Guangzhou RiboBio Co.,
Ltd. Cells (5×105) were seeded into 6-well plates and
then were transfected with 100 nM Cry1 siRNA at 37°C for 24 or 48 h
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Non-targeting
siRNA was used as control.
Senescence-associated β-galactosidase
(SA-β-Gal) cell staining
Senescent cells were detected using the SA-β-Gal
staining kit according to the manufacturer's instruction (Cell
Signaling Technology, Inc.). Cells were washed with PBS and fixed
with 4% paraformaldehyde at room temperature for 15 min. Each well
(6-well plates) was filled with 1 ml β-Gal staining solution. The
plate was sealed with a parafilm and incubated at 37°C overnight in
a dry incubator (no CO2). The percentage of
SA-β-Gal-positive cells were identified as bluish green-stained
cells under a fluorescent microscope (magnification, ×400; Nikon
Corporation).
Co-immunoprecipitation (Co-IP)
After washing in cold PBS, cells were lysed with
RIPA lysis buffer containing 1 mM PMSF (Fude) and phosphatase
inhibitor (Fude). Total cell lysates (1.0 ml) were incubated with 1
µg anti-p53 (cat. no. 2524; Cell Signaling Technology, Inc.)
overnight at 4°C, and then mixed with 40 µl protein-A + G
conjugated beads (Beyotime Institute of Biotechnology) to each
sample. Centrifuged at 4°C for 14,000 × g for 15 min. Beads were
then washed with lysis buffer and centrifugation was repeated three
times. The immunoprecipitated protein complexes were analyzed via
western blotting.
Statistical analysis
Statistical analysis was performed using SPSS 20.0
software (IBM Corp.). Differences between groups were compared
using one-way ANOVA followed by Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant difference.
In total, three duplicated experiment were performed.
Results
Res cells resist PTX-induced
senescence
To study drug resistance, Res UMUC3 and EJ cells
were established via prolonged culture in the presence of
cisplatin. As presented in Fig.
S1, the selected cells were resistant to cisplatin, as well as
PTX. Res cells exhibited normal proliferative profile as
demonstrated by results of DNA replication (Fig. 1A), cell cycle distribution (Fig. 1B) and the expression of cell
cycle-related proteins, including Cyclin D1, Cyclin E1 and
minichromosome maintenance complex component 7 (MCM7) (Fig. 1C). Upon acute PTX treatment, Res
cells presented with significantly decreased DNA replication
(Fig. 1A) and
G1/G0 accumulation (Fig. 1B). Moreover, the proteins expression
levels of Cyclin D1, Cyclin E1 and MCM7 were significantly
decreased (Fig. 1C). These data
indicated that the Res cells entered a non-proliferative quiescent
state upon PTX stress.
It has been reported that prolonged proliferation
arrest could lead to cell senescence (25) and that PTX can induce cell
senescence (16,17). In UMUC3 and EJ cells with a 2-day
PTX exposure, it was observed that the staining of SA-β-Gal was
20–50 times higher compared with that in cells without PTX
treatment (Fig. 1D). Accordingly,
the expression levels of the canonical senescence-associated
proteins, p53 and p21, were increased after PTX treatment; however,
p53 expression demonstrated no statistical difference after PTX
treatment in EJ cells (Fig. 1E).
While UMUC3 cells carry a homozygous tumor protein p53 mutation, it
has been revealed that p53 function still remains in UMUC3 cells
(26). However, in the UMUC3 Res
and EJ Res cells, nearly no SA-β-Gal positive signals were detected
after PTX treatment (Fig. 1D).
Consistently, p53 protein expression was decreased in PTX-treated
Res cells (Fig. 1E). p21 expression
was also decreased in EJ Res cells, but increased in the UMUC3 Res
cells, indicating the discrepancy between the different cell types
(Fig. 1E). According to the ATCC
and a previous report, the cyclin dependent kinase inhibitor 2A
(cdkn2a), a gene encoding p16 protein, is mutated or deleted in
UMUC3 and EJ cells (27,28). Consistent with this, no p16 protein
was detected in both cell lines (data not shown). These results
demonstrated that the Res cells appeared to escape from senescence
and remained in a quiescent state under PTX stress.
Res cells enter quiescence with
prolonged circadian rhythm
The circadian rhythm is closely associated with cell
proliferation (29), and also
regulates cell senescence (30).
The expression levels of the core circadian genes, BMAL1, CLOCK,
PER2 and CRY1, were detected in the Res cells. The mRNA expression
levels of the four core circadian genes fluctuated over a period of
24 h, similar with those in the parental cells (Fig. 2A). However, the circadian
oscillation was disturbed after PTX treatment demonstrated by an
increased amplitude and a prolonged period in the Res cells. In the
UMUC3 Res cells, the circadian period was ~48 h, and in the EJ Res
cells circadian oscillation was not noticed in a period of 48 h
(Fig. 2A). Moreover, the expression
of the circadian genes at the protein level showed similar
oscillation before and after PTX treatment. However, the amplitude
of CRY1 oscillation was reduced by PTX in both UMUC3 Res (Fig. 2B) and EJ Res cells (Fig. S2).
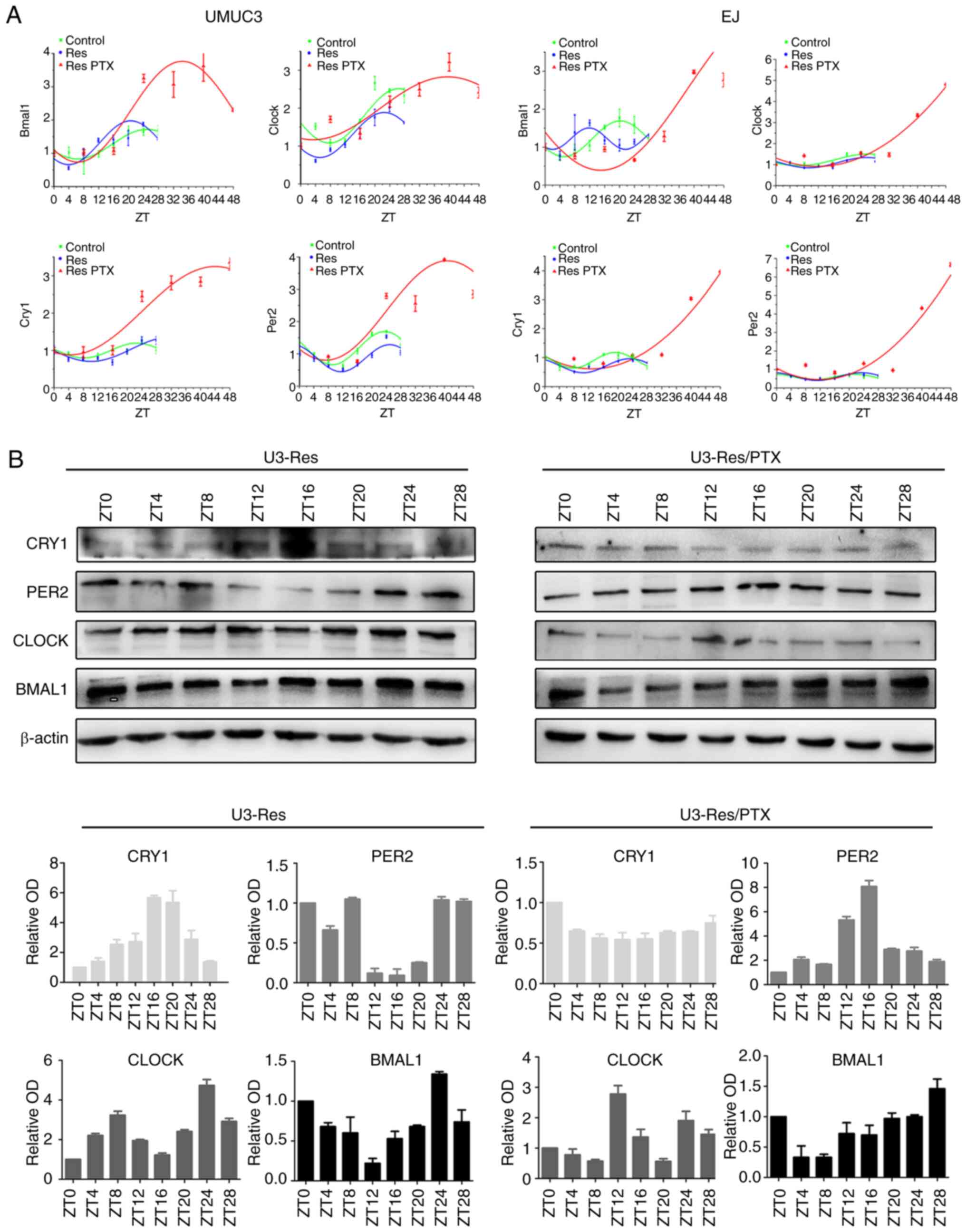 | Figure 2.Res cells enter quiescence with
prolonged circadian period. After cells (UMUC3 and EJ, Res and Res
+ PTX) were synchronized with 0.1 µM dexamethasone for 2 h,
circadian genes were detected at different time points. (A) mRNA
expression levels of circadian clock genes (BMAL1, CLOCK, PER2 and
CRY1) and corresponding fitted cosinor curves. (B) Time course of
protein (BMAL1, CLOCK, PER2 and CRY1) expression levels in the Res
and Res + PTX (80 nM) cells. β-Actin was used as loading control.
Data are presented as the OD fold difference related to the control
from three duplicate experiments. Res, cisplatin-resistant cells;
PTX, paclitaxel; OD, optical density; ZT, circadian time; CRY1,
Cryptochrome 1; PER2, period 2; CLOCK, circadian locomotor output
cycles kaput; BMAL1, brain and muscle Arnt-like protein 1. |
To further clarify the relationship between the
circadian rhythm and cell proliferation, the expression levels of
circadian gene were examined in serum-starved cells. As expected,
serum starvation induced a prolonged circadian oscillation
(Fig. S3). These results indicated
that the quiescent status of PTX-treated Res cells was accompanied
with a prolonged circadian rhythm.
CRY1 is accumulated in Res cells after
PTX treatment
Next, the expression levels of the four circadian
proteins were compared in the parental, Res and Res + PTX cells. In
UMUC3 cells, BMAL1, CLOCK and PER2 expression levels were decreased
in the Res and Res + PTX cells compared with parental UMUC3 cells,
but there was no significant difference between Res and Res + PTX
cells. Noticeably, CRY1 protein was significantly increased in the
Res + PTX cells (Fig. 3A). In EJ
cells, CRY1 and BMAL1 demonstrated similar trends as in UMUC3
cells. PER2 expression increased in the Res cells compared with
control and Res + PTX cells. CLOCK expression was significantly
increased in the Res + PTX group vs. Control and Res groups
(Fig. 3A). The circadian proteins
were further compared in the serum-starvation cells. After
starvation for 72 h, CRY1 and PER2 accumulated significantly in
both cell lines. Moreover, CLOCK and BMAL1 expression levels
decreased in UMUC-3 cells, but there were no significant changes in
EJ cells after serum-starvation for 72 h (Fig. 3B).
 | Figure 3.Expression levels of circadian clock
proteins in the Res cells with PTX treatment. (A) Total extracts of
cells were harvested at 2 h post-dexamethasone (0.1 µM). The
expression levels of β-Actin and circadian clock proteins,
including BMAL1, CLOCK, PER2 and CRY1, were assessed via western
blot analysis. (B) EJ and UMUC3 cells were treated with serum-free
medium for 24, 48 and 72 h. The expression levels of BMAL1, CLOCK,
PER2 and CRY1 were detected using western blotting. Data are
presented as the OD fold difference related to the control from
three duplicate experiments. *P<0.05. CRY1, Cryptochrome 1;
PER2, period 2; CLOCK, circadian locomotor output cycles kaput;
BMAL1, brain and muscle Arnt-like protein 1; Res,
cisplatin-resistant cells; PTX, paclitaxel; OD, optical
density. |
CRY1 knockdown induces senescence in
Res cells after PTX treatment
It was further examined whether CRY1 functioned as
an inhibitor in PTX-induced senescence. To test this, siRNA was
used to knockdown CRY1 expression in the PTX-treated Res cells.
(Fig. 4A and B). After CRY1
knockdown, p21 mRNA expression was significantly downregulated
(Fig. 4A), but its protein
expression was not significantly changed in both Res cells
(Fig. 4B). The p53 mRNA expression
did not change significantly in EJ Res cells, but was significantly
increased in UMUC3 Res cells (Fig.
4A). Moreover, its protein expression was significantly
enhanced after CRY1 knockdown in both cell lines (Fig. 4B). The si-CRY1-1 demonstrated a
higher efficiency compared with the si-CRY1-2 to inhibit CRY1
expression. Therefore, the si-CRY1-1 was used in subsequent
experiments. It was identified that CRY1 knockdown induced apparent
senescence in the PTX-treated Res cells (Fig. 4C). Furthermore, apoptosis was
significantly enhanced in the PTX-treated Res cells by CRY1
knockdown (Fig. 4D). These results
indicated that CRY1 contributed to resistance of senescence by
decreasing p53 protein expression.
CRY1 promotes MDM2 proto-oncogene
(MDM2)-mediated p53 degradation
The E3 ubiquitin ligase MDM2 mediates p53
ubiquitination, leading to p53 degradation, which is the major
mechanism for p53 protein turnover (31) Since CRY1 knockdown increased the
expression of p53 protein, but not p53 mRNA, it was investigated
whether CRY1 participated in MDM2-mediated p53 degradation in the
Res cells. The Co-IP assay using anti-p53 antibody successfully
detected an interaction between endogenous MDM2 and p53 proteins.
It was found that CRY1 knockdown significantly decreased MDM2
expression in the immunoprecipitation complex (Fig. 5), suggesting that the MDM2/p53
interaction was attenuated by CRY1 knockdown. This indicated that
CRY1 could facilitate the interaction of p53 with MDM2 and
potentiate p53 degradation.
Discussion
Cellular quiescence or dormancy is an evolutionary
conserved mechanism of survival, which helps cells adapt to stress
and survive a hostile environment (32). Glioblastoma cells can enter a
quiescent state to acquire survival advantages and ultimately
chemoresistance (33,34). Fluorouracil (5FU)-resistant human
colon cancer cells will enter a reversible quiescent G0-state upon
re-exposure to higher 5FU concentrations (35). Our previous research revealed that
cancer cells enter quiescence in acidic extracellular culture
(36). In the current study, when
the Res BC cells were treated with higher PTX, cells were arrested
at G1/G0, DNA replication was markedly
inhibited and, more importantly, the expression levels of
G1-S cyclin proteins (Cyclin D1 and E1) were
significantly decreased. Therefore, entering quiescence is an
effective mechanism for tumor cells to resist chemotherapy and
radiation in various types of cancer, and present as an important
mechanism of drug-resistance (37).
The present study identified a prolonged circadian
rhythm in quiescent cells, including PTX-treated Res cells and
serum-starved cells. Furthermore, CRY1 protein expression was found
to be increased in quiescent cells. CRY1 is a core clock repressor
that, along with PER, determines circadian periodicity (38), and stabilization of CRY1 prolongs
the circadian rhythm (39).
Therefore, the prolonged circadian rhythm may due to the
accumulation of CRY1 protein. It has been revealed that disruption
of the circadian rhythm could drive intrinsic resistance to
anticancer drugs, including PTX (40), Doxorubicin (41) and tamoxifen (42). More specifically, circadian proteins
are reported to regulate drug resistance. The gene timeless
circadian regulator confers cisplatin resistance and could
represent a valuable prognostic factor in nasopharyngeal carcinoma
(43). CLOCK regulates the
expression of Tip60 and contributes to cisplatin resistance
(44). KS15, an inhibitor of CRY,
decreases the speed of cell proliferation and increases the
sensitivity of MCF-7 cells to doxorubicin and tamoxifen (45). These results confirm a close
association between the circadian rhythm and drug resistance.
TIS is gaining increased attention in anticancer
therapy (46,47). The present study demonstrated that
PTX induced apparent senescence in BC cells, but not in the Res
cells. Since these Res cells were arrested in
G1/G0 phase under PTX stress, it is of great
important to investigate how these cells escape from senescence. It
was identified that the circadian protein CRY1 was accumulated in
these Res cells and, more importantly, CRY1 knockdown could restore
the ability of PTX to induce senescence. The current results
indicated that the accumulation of CRY1 protected BC cells against
PTX-induced senescence. On the contrary, it has been reported that
in mouse embryonic fibroblasts CRY1/2 functions as an inducer of
oncogene-induced senescence by suppressing the activating
transcription factor 4, a potent repressor of p16 and p19 (also
known as p14 in humans) (48).
However, this pathway may not function in BC cells, as cdkn2a (the
gene encoding p16 protein) is mutated or missing in these cells
(27). Moreover, such a
controversial phenotype may be explained by a different type of
senescence, such as therapy-vs. oncogene-induced senescence. The
difference between healthy fibroblasts and tumor cells may also
lead to different results (49).
Other circadian proteins have been shown to regulate cell
senescence. For instance, BMAL1KO mice present signs of epidermal
stem cells ageing, which is accompanied by increased expression of
p16 (50), while CLOCK mutant mice
have an accelerated aging program at low-dose irradiation (51). Collectively, these results highlight
the existence of a complex interconnection between senescence and
individual components of the circadian clock machinery.
The present study identified that CRY1 knockdown
increased p53 protein expression without changing p53 mRNA level,
indicating that CRY1 promoted p53 degradation. Furthermore, it was
demonstrated that CRY1 facilitated the interaction of p53 with
MDM2, contributing to p53 degradation. Consistently, it has been
reported previously that CRY1 could promote FOXO1 degradation by
promoting FOXO1 binding to the ubiquitin E3 ligase MDM2 (31). Thus, it is suggested that CRY1 acts
as a coordinator with MDM2. Similarly, another cryptochrome
protein, CRY2, has been identified as a component of F-box and
leucine rich repeat protein 3-containing E3 ligase that recruits
c-MYC for ubiquitination (52,53).
In addition, CRY1 can interact with the glucocorticoid receptor and
widely alter the transcriptional response to glucocorticoids in
mouse embryonic fibroblasts (54).
CRY can also increase insulin-like growth factor transcription by
promoting JAK2-dependent STAT5 phosphorylation (55). Taken together, these findings
suggested that, as a clock repressor, CRY1 also functioned to
connect circadian rhythms with multiple other physiological or
pathological processes.
In conclusion, the present study demonstrated an
inhibitory effect of CRY1 on PTX-induced cellular senescence in BC.
Thus, targeting CRY1 may be a feasible strategy to induce
senescence in cancer cells. However, the present study was limited
to PTX-induced senescence in BC, and it is important to investigate
whether CRY1 has similar anti-senescence function in other types of
cancer cells, and whether CRY1 still functions in other types of
senescence.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Junfang Ji
(Zhejiang University) and Dr Ziyi Wang (Zhejiang University) for
improving the English writing of this article.
Funding
This work was supported by the grants from the
Shenzhen Science and Technology Project (grant nos.
JCYJ20180305164655077 and JCYJ20180305124227251) and the National
Natural Science Foundation of China (grant no. 81672915).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and XL designed the experiments. MJ, BS, LM, WQ,
JY, PL and BY performed the experiments. MJ, DL, DW, LX, HL and ZZ
analyzed the experimental results. JL and MJ wrote the manuscript.
All authors reviewed the manuscript, and have read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Casadei C, Dizman N, Schepisi G, Cursano
MC, Basso U, Santini D, Pal SK and De Giorgi U: Targeted therapies
for advanced bladder cancer: New strategies with FGFR inhibitors.
Ther Adv Med Oncol. 11:17588359198902852019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alfred Witjes J, Lebret T, Compérat EM,
Cowan NC, De Santis M, Bruins HM, Hernández V, Espinós EL, Dunn J,
Rouanne M, et al: Updated 2016 EAU guidelines on muscle-invasive
and metastatic bladder cancer. Eur Urol. 71:462–475. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ke B, Wei T, Huang Y, Gong Y, Wu G, Liu J,
Chen X and Shi L: Interleukin-7 resensitizes non-small-cell lung
cancer to cisplatin via inhibition of ABCG2. Mediators Inflamm.
2019:72414182019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Li LJ, Qiu MX and Gong BS: Effects
of paclitaxel combined with miR-448 on growth and proliferation of
bladder cancer EJ cells. Eur Rev Med Pharmacol Sci. 22:3363–3369.
2018.PubMed/NCBI
|
|
5
|
Zeng Q, Liu J, Cao P, Li J, Liu X, Fan X,
Liu L, Cheng Y, Xiong W, Li J, et al: Inhibition of REDD1
sensitizes bladder urothelial carcinoma to paclitaxel by inhibiting
autophagy. Clin Cancer Res. 24:445–459. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He Q, Li J, Yin W, Song Z, Zhang Z, Yi T,
Tang J, Wu D, Lu Y, Wang Z, et al: Low-dose paclitaxel enhances the
anti-tumor efficacy of GM-CSF surface-modified whole-tumor-cell
vaccine in mouse model of prostate cancer. Cancer Immunol
Immunother. 60:715–730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Michaelson MD, Hu C, Pham HT, Dahl DM,
Lee-Wu C, Swanson GP, Vuky J, Lee RJ, Souhami L, Chang B, et al: A
phase 1/2 trial of a combination of paclitaxel and trastuzumab with
daily irradiation or paclitaxel alone with daily irradiation after
transurethral surgery for noncystectomy candidates with
muscle-invasive bladder cancer (Trial NRG Oncology RTOG 0524). Int
J Radiat Oncol Biol Phys. 97:995–1001. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Albers P, Park SI, Niegisch G, Fechner G,
Steiner U, Lehmann J, Heimbach D, Heidenreich A, Fimmers R and
Siener R; AUO Bladder Cancer Group, : Randomized phase III trial of
2nd line gemcitabine and paclitaxel chemotherapy in patients with
advanced bladder cancer: Short-term versus prolonged treatment
[German association of urological oncology (AUO) trial AB 20/99].
Ann Oncol. 22:288–294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mari A, D'Andrea D, Abufaraj M, Foerster
B, Kimura S and Shariat SF: Genetic determinants for chemo- and
radiotherapy resistance in bladder cancer. Transl Androl Urol.
6:1081–1089. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao YH, Qin XL, Yang JY, Liao YW, Wu XZ
and Zheng HP: Identification and expression analysis of ceftriaxone
resistance-related genes in Neisseria gonorrhoeae integrating
RNA-Seq data and qRT-PCR validation. J Glob Antimicrob Resist.
16:202–209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang S, Xie J, Miao J, Li R, Liao W and
Luo R: A knockdown of Maml1 that results in melanoma cell
senescence promotes an innate and adaptive immune response. Cancer
Immunol Immunother. 62:183–190. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mikula-Pietrasik J, Niklas A, Uruski P,
Tykarski A and Książek K: Mechanisms and significance of
therapy-induced and spontaneous senescence of cancer cells. Cell
Mol Life Sci. 77:213–229. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Collado M and Serrano M: Senescence in
tumours: Evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee S and Lee JS: Cellular senescence: A
promising strategy for cancer therapy. BMB Rep. 52:35–41. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin S, Schulte BA and Wang GY: Role of
senescence induction in cancer treatment. World J Clin Oncol.
9:180–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khongkow P, Gomes AR, Gong C, Man EP,
Tsang JW, Zhao F, Monteiro LJ, Coombes RC, Medema RH, Khoo US and
Lam EW: Paclitaxel targets FOXM1 to regulate KIF20A in mitotic
catastrophe and breast cancer paclitaxel resistance. Oncogene.
35:990–1002. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kavanagh EL, Lindsay S, Halasz M, Gubbins
LC, Weiner-Gorzel K, Guang MHZ, McGoldrick A, Collins E, Henry M,
Blanco-Fernández A, et al: Protein and chemotherapy profiling of
extracellular vesicles harvested from therapeutic induced senescent
triple negative breast cancer cells. Oncogenesis. 6:e3882017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baba K and Tosini G: Aging alters
circadian rhythms in the mouse eye. J Biol Rhythms. 33:441–445.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rakshit K, Wambua R, Giebultowicz TM and
Giebultowicz JM: Effects of exercise on circadian rhythms and
mobility in aging Drosophila melanogaster. Exp Gerontol.
48:1260–1265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takahashi JS: Transcriptional architecture
of the mammalian circadian clock. Nat Rev Genet. 18:164–179. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Relles D, Sendecki J, Chipitsyna G, Hyslop
T, Yeo CJ and Arafat HA: Circadian gene expression and
clinicopathologic correlates in pancreatic cancer. J Gastrointest
Surg. 17:443–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taniguchi H, Fernández AF, Setién F,
Ropero S, Ballestar E, Villanueva A, Yamamoto H, Imai K, Shinomura
Y and Esteller M: Epigenetic inactivation of the circadian clock
gene BMAL1 in hematologic malignancies. Cancer Res. 69:8447–8454.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu Y, Stevens RG, Hoffman AE, Fitzgerald
LM, Kwon EM, Ostrander EA, Davis S, Zheng T and Stanford JL:
Testing the circadian gene hypothesis in prostate cancer: A
population-based case-control study. Cancer Res. 69:9315–9322.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Terzi MY, Izmirli M and Gogebakan B: The
cell fate: Senescence or quiescence. Mol Biol Rep. 43:1213–1220.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang L, Li Y, Bhattacharya A and Zhang Y:
PEPD is a pivotal regulator of p53 tumor suppressor. Nat Commun.
8:20522017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grim J, D'Amico A, Frizelle S, Zhou J,
Kratzke RA and Curiel DT: Adenovirus-mediated delivery of p16 to
p16-deficient human bladder cancer cells confers chemoresistance to
cisplatin and paclitaxel. Clin Cancer Res. 3:2415–2423.
1997.PubMed/NCBI
|
|
28
|
Sanchez-Carbayo M, Socci ND, Charytonowicz
E, Lu M, Prystowsky M, Childs G and Cordon-Cardo C: Molecular
profiling of bladder cancer using cDNA microarrays: Defining
histogenesis and biological phenotypes. Cancer Res. 62:6973–6980.
2002.PubMed/NCBI
|
|
29
|
Gaucher J, Montellier E and Sassone-Corsi
P: Molecular cogs: Interplay between circadian clock and cell
cycle. Trends Cell Biol. 28:368–379. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sulli G, Rommel A, Wang X, Kolar MJ, Puca
F, Saghatelian A, Plikus MV, Verma IM and Panda S: Pharmacological
activation of REV-ERBs is lethal in cancer and oncogene-induced
senescence. Nature. 553:351–355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jang H, Lee GY, Selby CP, Lee G, Jeon YG,
Lee JH, Cheng KK, Titchenell P, Birnbaum MJ, Xu A, et al:
SREBP1c-CRY1 signalling represses hepatic glucose production by
promoting FOXO1 degradation during refeeding. Nat Commun.
7:121802016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Manjili MH: Tumor dormancy and relapse:
From a natural byproduct of evolution to a disease state. Cancer
Res. 77:2564–2569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang L, Shang Z, Zhou Y, Hu X, Chen Y, Fan
Y, Wei X, Wu L, Liang Q, Zhang J and Gao Z: Autophagy mediates
glucose starvation-induced glioblastoma cell quiescence and
chemoresistance through coordinating cell metabolism, cell cycle,
and survival. Cell Death Dis. 9:2132018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Atkins RJ, Stylli SS, Kurganovs N,
Mangiola S, Nowell CJ, Ware TM, Corcoran NM, Brown DV, Kaye AH,
Morokoff A, et al: Cell quiescence correlates with enhanced
glioblastoma cell invasion and cytotoxic resistance. Exp Cell Res.
374:353–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Touil Y, Igoudjil W, Corvaisier M, Dessein
AF, Vandomme J, Monté D, Stechly L, Skrypek N, Langlois C, Grard G,
et al: Colon cancer cells escape 5FU chemotherapy-induced cell
death by entering stemness and quiescence associated with the
c-Yes/YAP axis. Clin Cancer Res. 20:837–846. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang YJ, Xu LL, Wang P, Jian H, Shi X,
Jia M, Mo L, Hu Z, Li H and Li J: Phenotypic transition of tumor
cells between epithelial- and mesenchymal-like state during
adaptation to acidosis. Cell Cycle. 18:1938–1947. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Angelis ML, Francescangeli F, La Torre
F and Zeuner A: Stem cell plasticity and dormancy in the
development of cancer therapy resistance. Front Oncol. 9:6262019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Toledo M, Batista-Gonzalez A, Merheb E,
Aoun ML, Tarabra E, Feng D, Sarparanta J, Merlo P, Botrè F,
Schwartz GJ, et al: Autophagy regulates the liver clock and glucose
metabolism by degrading CRY1. Cell Metab. 28:268–281.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hirota T, Lee JW, St John PC, Sawa M,
Iwaisako K, Noguchi T, Pongsawakul PY, Sonntag T, Welsh DK, Brenner
DA, et al: Identification of small molecule activators of
cryptochrome. Science. 337:1094–1097. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xiang S, Dauchy RT, Hoffman AE, Pointer D,
Frasch T, Blask DE and Hill SM: Epigenetic inhibition of the tumor
suppressor ARHI by light at night-induced circadian melatonin
disruption mediates STAT3-driven paclitaxel resistance in breast
cancer. J Pineal Res. 67:e125862019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiang S, Dauchy RT, Hauch A, Mao L, Yuan
L, Wren MA, Belancio VP, Mondal D, Frasch T, Blask DE and Hill SM:
Doxorubicin resistance in breast cancer is driven by light at
night-induced disruption of the circadian melatonin signal. J
Pineal Res. 59:60–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dauchy RT, Xiang S, Mao L, Brimer S, Wren
MA, Yuan L, Anbalagan M, Hauch A, Frasch T, Rowan BG, et al:
Circadian and melatonin disruption by exposure to light at night
drives intrinsic resistance to tamoxifen therapy in breast cancer.
Cancer Res. 74:4099–4110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu SL, Lin HX, Lin CY, Sun XQ, Ye LP, Qiu
F, Wen W, Hua X, Wu XQ, Li J, et al: TIMELESS confers cisplatin
resistance in nasopharyngeal carcinoma by activating the
Wnt/β-catenin signaling pathway and promoting the epithelial
mesenchymal transition. Cancer Lett. 402:117–130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miyamoto N, Izumi H, Noguchi T, Nakajima
Y, Ohmiya Y, Shiota M, Kidani A, Tawara A and Kohno K: Tip60 is
regulated by circadian transcription factor clock and is involved
in cisplatin resistance. J Biol Chem. 283:18218–18226. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chun SK, Chung S, Kim HD, Lee JH, Jang J,
Kim J, Kim D, Son GH, Oh YJ, Suh YG, et al: A synthetic
cryptochrome inhibitor induces anti-proliferative effects and
increases chemosensitivity in human breast cancer cells. Biochem
Biophys Res Commun. 467:441–446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chang BD, Xuan Y, Broude EV, Zhu H, Schott
B, Fang J and Roninson IB: Role of p53 and p21waf1/cip1 in
senescence-like terminal proliferation arrest induced in human
tumor cells by chemotherapeutic drugs. Oncogene. 18:4808–4818.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang BD, Broude EV, Dokmanovic M, Zhu H,
Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K and Roninson IB: A
senescence-like phenotype distinguishes tumor cells that undergo
terminal proliferation arrest after exposure to anticancer agents.
Cancer Res. 59:3761–3767. 1999.PubMed/NCBI
|
|
48
|
Katamune C, Koyanagi S, Shiromizu S,
Matsunaga N, Shimba S, Shibata S and Ohdo S: Different roles of
negative and positive components of the circadian clock in
oncogene-induced neoplastic transformation. J Biol Chem.
291:10541–10550. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wyld L, Bellantuono I, Tchkonia T, Morgan
J, Turner O, Foss F, George J, Danson S and Kirkland JL: Senescence
and cancer: A review of clinical implications of senescence and
senotherapies. Cancers (Basel). 12:21342020. View Article : Google Scholar
|
|
50
|
Janich P, Pascual G, Merlos-Suárez A,
Batlle E, Ripperger J, Albrecht U, Cheng HY, Obrietan K, Di Croce L
and Benitah SA: The circadian molecular clock creates epidermal
stem cell heterogeneity. Nature. 480:209–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Antoch MP, Gorbacheva VY, Vykhovanets O,
Toshkov IA, Kondratov RV, Kondratova AA, Lee C and Nikitin AY:
Disruption of the circadian clock due to the Clock mutation has
discrete effects on aging and carcinogenesis. Cell Cycle.
7:1197–1204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huber AL, Papp SJ, Chan AB, Henriksson E,
Jordan SD, Kriebs A, Nguyen M, Wallace M, Li Z, Metallo CM and
Lamia KA: CRY2 and FBXL3 cooperatively degrade c-MYC. Mol Cell.
64:774–789. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Du H, Jie L, Xu W, Wu Y, Liu T and Li M: A
monoclonal antibody against a potential cancer biomarker, human
ubiquitin-conjugating enzyme E2. Hybridoma (Larchmt). 31:196–202.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lamia KA, Papp SJ, Yu RT, Barish GD,
Uhlenhaut NH, Jonker JW, Downes M and Evans RM: Cryptochromes
mediate rhythmic repression of the glucocorticoid receptor. Nature.
480:552–556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chaudhari A, Gupta R, Patel S, Velingkaar
N and Kondratov R: Cryptochromes regulate IGF-1 production and
signaling through control of JAK2-dependent STAT5B phosphorylation.
Mol Biol Cell. 28:834–842. 2017. View Article : Google Scholar : PubMed/NCBI
|

















