Introduction
The viral family Flaviviridae includes the
genera Flavivirus, Pestivirus, Pegivirus and
Hepacivirus. Arthropod vectors, mainly ticks and mosquitoes,
constitute the transmission pathway of Flaviviridae viruses,
causing epidemics and medical concerns due to the large number of
diseases that they inflict on both humans and animals (1,2). The
different members of the Flaviviridae family share some
common elements of viral organization. Their viral particles
(virions) are small (~50 nm), spherical and enveloped, that
incorporate a single-stranded RNA of 9.5-12.5 kb (1). The viral genome is located inside the
capsid of the virion, having a positive-sense polarity and a long
open reading frame, which is flanked by untranslated regions at the
5' and 3' ends. The translated polyprotein consists of three
structural [capsid (C), membrane (M) and envelope (E)] and seven
non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5).
The latter region of NS proteins, at the C-terminal part of the
polyprotein, has a great contribution in the RNA replication
process. The most crucial NS proteins are the viral helicase and
the viral RNA-dependent RNA polymerase (RdRp) (2). The viral helicase constitutes the
main subject in the present study.
The viral RNA helicases are attributed to the NS3
region of the viral polyprotein, and they are involved in duplex
unwinding, during viral RNA replication. Being a promising
antiviral target, helicase inhibition leads to the pause of the
replication, proliferation and consequently, to the survival and
transmission of Flaviviridae viruses (3). Spondweni virus (SPONV) is a member of
the Flaviviridae virus family, of the genus
Flavivirus, and belongs to a serogroup with the Zika virus.
SPONV was first detected in Nigeria and South Africa in the 1950s
and subsequently, in sub-Saharan Africa. The viral cycle is located
between mosquitoes and non-human primates, causing symptomatic
infections of mild illness. However, some cases result in more
severe disease, including febrile syndrome, Vascular leakage
(shock) or neurological impairments. The epidemic of Zika virus
that broke out in the Western Hemisphere, reveals the epidemic
potential of SPONV and the increased number of infections in the
near future (4,5).
To date, there is no anti-SPONV therapy available,
while at the same time, the continued threat of emerging
Flavivirus remains incurable. This condition highlights the
need for an extensive fundamental study of viral biology, in order
to develop novel antiviral vaccines and drugs. The present study
proposes the three-dimensional structure of the helicase/protease
enzyme of SPONV, since its 3D structure has not yet been resolved.
The structure is modelled by applying homology modelling techniques
and using the crystal structure of the Dengue virus-4 (DENV-4)
helicase/protease (PDB: 2VBC), of the same viral family
(Flaviviridae), as the template (6). The next challenge of the study was
the confirmation testing of the functionality, efficacy and
reliability of the model in structure-based drug design strategies.
For this purpose, the resolved structure of the hepatitis C virus
(HCV) helicase (PDB: 1A1V) was used, as it is complexed with a
single-stranded RNA, a key molecule for the establishment of
interactions with a future inhibitor of the SPONV helicase
(7).
Materials and methods
Database sequence search
The amino acid sequence data of the NS3 helicase of
the Flaviviridae family were collected from the NCBI
database, by using related keywords and Virus Pathogen Database and
Analysis Resource (ViPR) (8,9). The
amino acid sequence of the SPONV NS3 protein was obtained from the
NCBI database, with the accession no. YP_009227191.1 and entry
name, Spondweni virus, non-structural protein NS3. The sequence
length is 619 aa. All the available NS3 viral protein sequences
were filtered in order to remove the irrelevant sequences, as well
as the hypothetical, partial and synthetic sequences. Moreover, the
final dataset of the NS3 viral proteins was merged using the
dataset from previous research, as previously described by
Papageorgiou et al (1).
Multiple sequence alignment (MSA) and
conserved motifs
MSA was executed using the MATLAB Bioinformatics
toolbox (https://uk.mathworks.com/products/bioinfo.html),
utilizing a guide tree and the progressive MSA method as previously
described (10,11). Pairwise distances among sequences
were estimated based on the pairwise alignment with the ‘Gonnet’
method and followed by calculating the differences between each
pair of sequences (12). The
Neighbor-Joining method was used towards to estimating the guide
tree by assuming equal variance and independence of evolutionary
distance estimates (13). Finally,
the major conserved helicase motifs, which are characteristic of
the Flaviviridae family, were identified using the consensus
sequences from the MSA. The visualization was performed using the
Jalview program, providing important information on sequence
conservation, quality and consensus (14).
Phylogenetic analysis
The construction of a phylogenetic tree is a
consequence of the MSA. For the purposes of the present study, the
identification of the homologous viral structure that will be used
as a template for homology modeling, a phylogenetic tree was
constructed with representatives of the genus Flavivirus.
The formation of separate groups among members of the genus
Flavivirus helps to identify which virus is the most closely
related to SPONV. The method that was used to create a phylogenetic
tree was the Neighbor-Joining method and the scores were computed
with the scoring matrix BLOSUM62. The visualization of the tree was
completed using the Jalview program (14-16).
Template identification
The BLASTp algorithm was used to identify the
template structure for the homology modeling procedure of SPONV
helicase/protease, by searching the Protein Data Bank (PDB)
(17,18). The most important alignments
emerged and according to the identity percentage and the crystal
resolution of the structures, the structure that will be the
template can be distinguished. Subsequently, another search was
performed through the Molecular Operating Environment (MOE) program
(www.chemcomp.com) against the PDB.
Homology modelling
Homology modelling of the SPONV NS3 helicase was
performed using MOE version 2016.0801 and its homology modelling
application (19-21).
All calculations and visual constructions were performed using this
program. The MOE homology model method is separated into four main
steps (22). First, a primary
fragment geometry specification. Second, the insertion and deletions
task. The third step is the loop selection and the side-chain
packing and the last step is the final model selection and
refinement.
Model evaluation
The evaluation of the quality and reliability of the
produced SPONV helicase model is vital for the viability of the
present study. As a result, the created model was evaluated within
the MOE package. For this purpose, the RMSD and
Geometry-Ramachandran Plot diagrams were calculated and their
contribution to the structural estimation of the protein's quality
is crucial (23,24). The RMSD calculation helps to
understand the quality of the system, while high RMSD values
indicate poor quality systems as opposed to low values, which are
indicative of good quality systems. Moreover, the Ramachandran plot
is the most reliable in silico tool for the enzyme's
stereochemical evaluation, looking at its phi/psi dihedral angles.
Last but not least, in order to analyze the molecular surface of
the produced SPONV helicase model, the electrostatic potential
surface was calculated by solving the non-linear Poisson-Boltzmann
equation using MOE software (20).
Identification of key amino acid
residues as candidate targets
A structural analysis was performed using the SPONV
model and the structural features from other available 3D
structures in an effort to confirm the functionality, suitability
and reliability of the SPONV helicase model. The HCV helicase (PDB:
1A1V) was the studied crystal structure, since several
structural-based drug design studies have been performed using this
crystal structure and they provide beneficial knowledge and a
number of pharmacophore models and inhibitors that have been
already designed (25-27).
Last but not least, some important conserved residues are isolated
and selected as a suitable target for structure-based experiments.
Based on this strategy, the present study attempted to exploit the
existing inhibitor molecules of HCV in order to inhibit the
activity of SPONV virus helicase.
Results and Discussion
MSA and phylogenetic analysis
The NS3 domain of Flaviviridae consists of
both the protease and the helicase coding regions (1). MSA of the NS3 protein sequences
indicate conservation in known and unknown important regions within
all Flaviviridae viruses, throughout the whole length of the
sequence (2). It is worth
mentioning that the NS3 sequences of the genus Pestivirus,
include an extended insertion at the N-terminal half of the
protein, which differentiates their size from Flavivirus,
Hepacivirus and Pegivirus. The conserved regions in
the four district genera indicate the several critical functional
domains of the enzyme. Based on the results from the extracted
consensus sequence of the NS3 MSA, all known conserved regions were
identified and highlighted (Fig.
1). Phylogenetic analysis of the representative NS3 viral
protein sequences indicates a clear separation between the genera
Flavivirus, Hepacivirus, Pestivirus and
Pegivirus of the Flaviviridae family (1). In the present study, the phylogenetic
tree was performed for the genus Flavivirus, in which SPONV
belongs (Fig. 2). The SPONV NS3
viral protein was identified in the same monophyletic branch with
Zika virus, and DENV-4.
The BLASTp algorithm was identified several protein
structures as a candidate protein templates in order to perform the
homology modelling of the NS3 viral protein of the SPONV. Two
parameters have been studied for the optimal selection of the final
template including i) the sequence identity; and ii) the crystal
structure resolution (2,16). The selected template was the
crystal structure of the NS3 protease/helicase from DENV-4 (PDB:
2VBC) (6). The DENV-4 belongs to
the same viral family (Flaviviridae) and genus
(Flavivirus). The 2VBC DENV-4 protease/helicase structure
has been established by X-ray crystallography at a 3.15 Å
resolution, having an identity percentage of 65,59%, a query cover
of 100% and a sequence length of 618 aa (6). Moreover, the crystal structure of the
HCV NS3 helicase, which has been co-crystalized with a
single-stranded DNA molecule (ssDNA), at a resolution of 2.2 Å
(PDB: 1A1V), has provided further information of the functionality
of the NS3 viral protein (7). HCV
belongs to the same family as SPONV (Flaviviridae) (1). The selection was made due to the
presence of ssDNA, contained in the crystal, but also due to the
extensive study of the structure for potential pharmacological
targets. Furthermore, all the available results and findings, as
described in the study by Vlachakis et al (3), for a possible pharmacophore model
against the viral NS3 protein of the Classical Swine Fever virus,
were studied. Classical swine fever virus is also a member of the
genus Flavivirus.
A MSA was constructed, including the SPONV
helicase/protease sequence, the DENV-4 helicase/protease sequence
(2VBC) and the HCV helicase sequence (1A1V) (Fig. 3). The alignment revealed the seven
major conserved motifs, which are characteristic and unique to the
helicases of the Flaviviridae viral family (motifs 1-6) and
these are highlighted in Fig. 2.
Moreover, in the MSA identified certain ‘key’ amino acids that are
necessary for the NS3 viral protein inhibition, as described in the
study by Vlachakis et al (3).
Description of the SPONV helicase
model
The candidate model of the SPONV helicase/protease
was established using the homology modeling process of the MOE
package (Fig. 4). Although the
Zika virus belongs to the same antigenic group with SPONV, and
consequently they have a similar helicase structure, the selected
template belongs to Dengue virus due to a greater query cover with
100% identity in sequence length, a relative higher identity
percentage throughout the protein sequence and a good-quality
crystal structure resolution (3.15 Å). Summarizing the results from
the MSA and the model, in the SPONV helicase/protease model, all
the structural features of known Flaviviridae helicases were
identified (Figs. 2 and 4). The model shares a similar structural
topology to its template (2VBC) (Fig.
4). Particularly, in the SPONV helicase/protease model, the
three distinct domains of helicases were structurally conserved, as
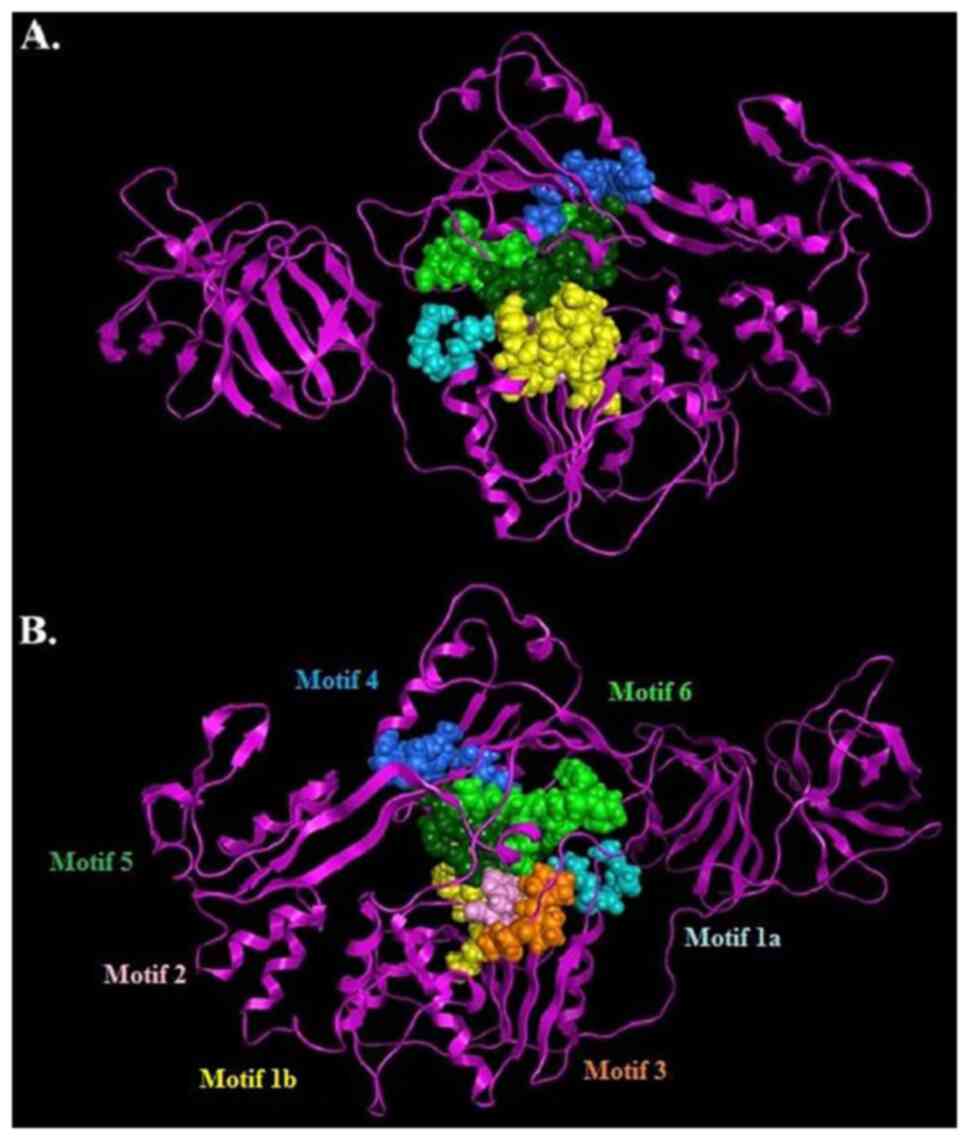
well as the protease region and various motifs (6). The seven characteristic motifs of the
SPONV helicase appeared in domains 1 and 2, exhibiting a connection
with NTP binding and hydrolysis (Fig.
5) (1,2,6,7). One
of the most vital motifs in Flaviviridae helicases is the
GxGKT/S Motif 1 in domain 1, which is conserved to the same loop in
kinases. It is also known as a Walker A motif, and it plays a
crucial role in the binding of β-phosphate of ATP (28,29).
Based on previous research, the mutagenesis within that motif
suggests that the mutant helicase is inactive (29). Furthermore, another important motif
for the helicase is the DExH Motif 2, in domain 1. The DExH Motif 2
is responsible for the binding of the Mg2+-ATP
substrate, establishing the optimum orientation of ATP for
nucleophilic attack (30-32).
Finally, although the crucial QRxGRxGR Motif 6 (in domain 2) is not
associated with ATP hydrolysis, and its function is exceptionally
crucial to the Flaviviridae helicase, as it is involved in
nucleic acid binding (7) (Fig. 5).
The SPONV helicase/protease model, same as all the
Flaviviridae helicases, consists of three helicase domains,
which are separated by two channels and placed at the C-terminal
part of the protein (2). The
domains 1 and 2 interact together and to a lesser extent with
domain 3. Domain 2 undergoes significant movements compared to the
other two domains, during the process of unwinding of
double-stranded nucleic acids. The channel between domain 3 and 1-2
accommodates ssRNA during the viral unwinding. RNA binds to the
helicase at the arginine-rich site of the 2nd domain (33). Moreover, significant interactions
have been identified in the conserved motifs between domains 1 and
2. Motif 3 is necessary in order to stabilize domains 1 and 2. At
the same direction, motif 7 forms critical contacts with motifs 1,
2, and 3(2). Last but not least,
the protease domain is located in the N-terminal of the model
(Figs. 4 and 5).
The model of SPONV was structurally superposed and
subsequently compared to its template and with the HCV crystal
structure (PDB: 1A1V) (Fig. 6).
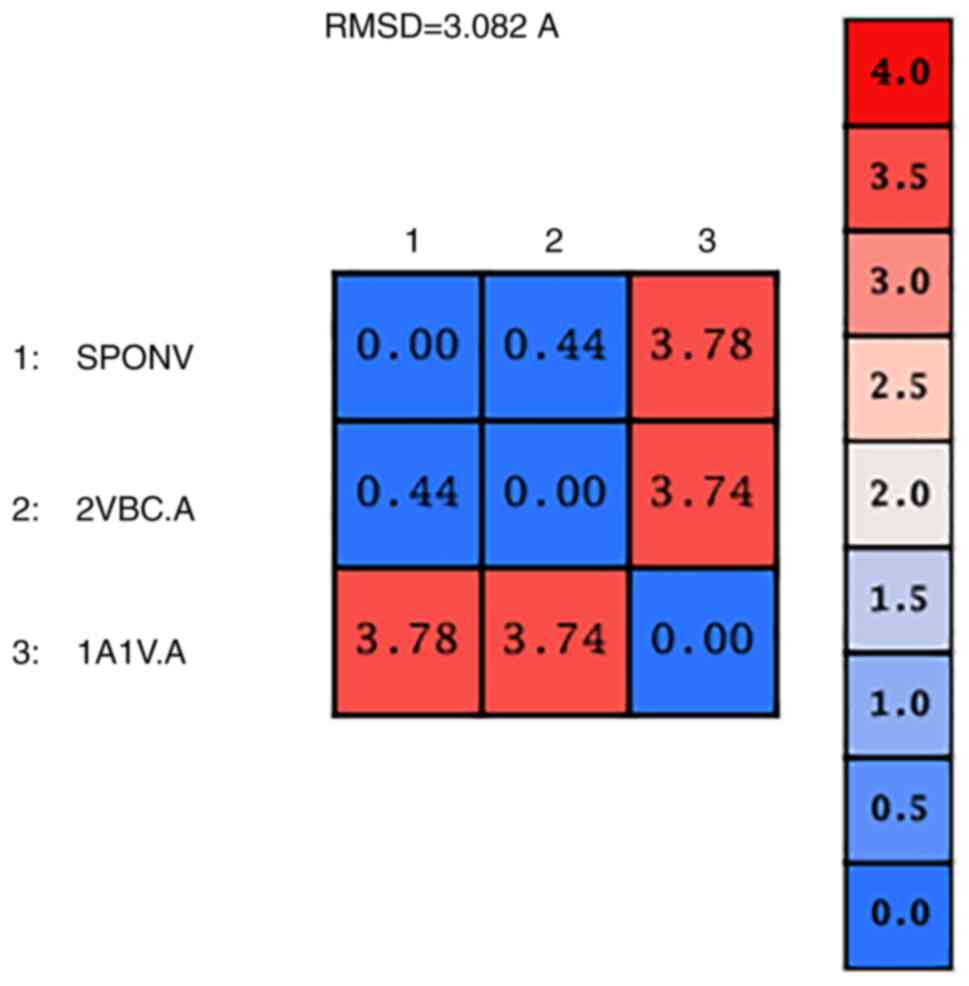
The alpha-carbon overall RMSD value between the two structures is
0.44 angstroms (Fig. 7). This
result confirms the similar configuration of the structures and the
good quality of the sequence alignment. However, the RMSD from the
superposition of the SPONV model and HCV crystal structure (1A1V)
was 3.78 angstroms (Fig. 7).
Although RMSD values in the range of RMSD ≤2 indicate unrelated
structures, in this case, the value is out of the optimal range due
to the lack of the protease domain in the 1A1V structure (Fig. 6). The fact automatically makes the
structures non-identical for this specific part of the protein.
Moreover, the three viruses, SPONV, DENV, HCV, share similar
helicase topology based on the secondary structure features, and a
very common active site, as all they belong to the
Flaviviridae family. The evaluation of the NS3 SPONV model
was performed using the Ramachandran method and the extracted
result is provided (Fig. 8)
(34). Three amino acids were
identified in disallowed regions, due to steric hindrance. A
structural analysis was performed in the MOE package in order to
examine those amino acids, and the results revealed that they
correspond to non-steric hindrance, alpha-helical and beta-sheet
conformations. The NS3 SPONV model was also examined for its
molecular and electrostatic potential surface (Fig. 9) (35). In the outcome, the results
represent the prediction of electrostatically preferred locations
of hydrophobic (colored white), H-bond acceptor (colored red) and
H-bond donor locations (colored blue). The graphical representation
of the electrostatic map provides additional information of the
proteins, including potential protein interactions (Fig. 9).
Key residues for the design of
candidate antiviral agents
The HCV helicase structure 1A1V was used as a
starting point in order to identify candidate key residues for the
inhibition of the NS3 SPONV helicase. Therefore, the ssDNA molecule
was transferred from the HCV structure into the produced NS3 SPONV
model, using structural superposition techniques as the MOE. Based
on findings from several studies, the channel between domain 3 and
1-2 is a possible candidate inhibition place, since it accommodates
the ssRNA during the viral unwinding and consequently plays a
critical role in the process of viral replication (3,6,7).
Conserved important residues which are necessary for
the helicase activity have been previously identified and have been
used as candidate targets for the design of antiviral agents
(36,37). Most of these are identified in
three important regions of the viral helicase, including Cys431,
Arg393 and Arg481. The first region with critical amino acids is
the ATP, a pocket-like hydrolysis active site. This region contains
an aspartic acid residue, which coordinates the hydrolysis of ATP
through its interaction with Mg2+ atoms (3,37).
Another important region which may contains key amino acid residues
is defined as the core of the domain 3 of Flaviviridae
helicases, where the anti-parallel β-sheets impart stability and
rigidity, key properties for the functional site of the enzyme.
Critical amino acid residues may also exist in the domain, between
domain 1 and 2 of the helicase, which constitutes the entrance site
of the incoming ssDNA, during the unwinding process (3,36).
Therefore, there is increasing interest for the design of candidate
inhibitors that will strongly interact with those key residues and
regions to block the ssRNA from the entrance into the helicase
channel.
Maintaining this strategy, the critical helicase
conserved ‘key’ residues were identified in the model as potential
targets for structure-based drug design. Using both previous
alignment and structural superposition, the residues of Cys429 and
Arg388 were identified at the corresponding positions (Figs. 3 and 10). It is obvious that Cys429 residue is
a key candidate pharmacological target, being directly exposed to
the solvent and strategically located at the center of the viral
helicase ssRNA channel. The Cysteine residue should create an S-S
or an S-C bond with the inhibitor molecule and to interact with
arginine residue, while the Arginine residue is expected to
establish H-bond with the inhibitor. The purpose is to develop a
bridge between the two conserved residues, Cys429 and Arg388, and
also to create a compound that strongly interacts with them,
blocking the passage of the ssRNA and consequently the helicase's
operation.
In conclusion, computational biology methods have
successfully bridged the gap between the lack of experimentally
determined protein structures and the design of antivirals,
allowing the prediction of a protein's three-dimensional
conformation in silico. The three-dimensional structure of
the SPONV helicase/protease model was designed using homology
modelling techniques. The template structure was the homologous
X-ray crystal structure of the DENV-4 helicase/protease (PDB entry:
2VBC), of the same Flaviviridae family. The evaluation of
the generated model was successful, exhibiting topological identity
to its template. The extensive study of the SPONV helicase/protease
model and the identification of key residues provide insight for
further studies. In silico methodologies have proven to be
an integral part of helicase inhibitor design. The latest Zika
outbreak and the ongoing COVID-19 pandemic have highlighted the
impact that viral infections can have on global communities and
health systems. Therefore, a proactive stance is imperative and the
modeling of structures of viral protein targets, such as the
Spondweni helicase through computational methods can enable
the design of potent antivirals.
Acknowledgements
Not applicable.
Funding
Funding: The authors would like to acknowledge funding from the
following organizations: i) AdjustEBOVGP-Dx (RIA2018EF-2081):
Biochemical Adjustments of native EBOV Glycoprotein in Patient
Sample to Unmask target Epitopes for Rapid Diagnostic Testing. A
European and Developing Countries Clinical Trials Partnership
(EDCTP2) under the Horizon 2020 ‘Research and Innovation Actions’
DESCA; and ii) ‘MilkSafe: A novel pipeline to enrich formula milk
using omics technologies’, a research co-financed by the European
Regional Development Fund of the European Union and Greek national
funds through the Operational Program Competitiveness,
Entrepreneurship and Innovation, under the call
RESEARCH-CREATE-INNOVATE (project code: T2EDK-02222).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors (LP, ET, EP, KID, KP, KD, DAS, FB, GPC,
EE and DV) contributed to the conceptualization and design of the
study, as well as in the writing, drafting, revising, editing and
reviewing of the manuscript. All authors confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Managing Editor of the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
GPC is an Editorial Advisor of the journal, but had no personal
involvement in the reviewing process, or any influence in terms of
adjudicating on the final decision, for this article. The other
authors declare that they have no competing interests.
References
|
1
|
Papageorgiou L, Loukatou S, Sofia K,
Maroulis D and Vlachakis D: An updated evolutionary study of
Flaviviridae NS3 helicase and NS5 RNA-dependent RNA polymerase
reveals novel invariable motifs as potential pharmacological
targets. Mol BioSyst. 12:2080–2093. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Papageorgiou L, Loukatou S, Koumandou VL,
Makałowski W, Megalooikonomou V, Vlachakis D and Kossida S:
Structural models for the design of novel antiviral agents against
greek goat encephalitis. PeerJ. 2(e664)2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Vlachakis D and Kossida S: Molecular
modeling and pharmacophore elucidation study of the classical swine
fever virus helicase as a promising pharmacological target. PeerJ.
1(e85)2013.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
White SK, Lednicky JA, Okech BA, Morris JG
and Dunford JC: Spondweni virus in field-caught culex
quinquefasciatus mosquitoes, Haiti, 2016. Emerg Infect Dis.
24:1765–1767. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pierson TC and Diamond MS: The continued
threat of emerging flaviviruses. Nat Microbiol. 5:796–812.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Luo D, Xu T, Hunke C, Grüber G, Vasudevan
SG and Lescar J: Crystal structure of the NS3 protease-helicase
from dengue virus. J Virol. 82:173–183. 2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kim JL, Morgenstern KA, Griffith JP, Dwyer
MD, Thomson JA, Murcko MA, Lin C and Caron PR: Hepatitis C virus
NS3 RNA helicase domain with a bound oligonucleotide: The crystal
structure provides insights into the mode of unwinding. Structure.
6:89–100. 1998.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pickett BE, Sadat EL, Zhang Y, Noronha JM,
Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, et al: ViPR:
An open bioinformatics database and analysis resource for virology
research. Nucleic Acids Res. 40:D593–D598. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
NCBI Resource Coordinators. Database
resources of the national center for biotechnology information.
Nucleic Acids Res. 44:D7–D19. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vlachakis D, Papageorgiou L, Papadaki A,
Georga M, Kossida S and Eliopoulos E: An updated evolutionary study
of the notch family reveals a new ancient origin and novel
invariable motifs as potential pharmacological targets. PeerJ.
8(e10334)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Papageorgiou L, Shalzi L, Pierouli K,
Papakonstantinou E, Manias S, Dragoumani K, Nicolaides NC,
Giannakakis A, Bacopoulou F, Chrousos GP, et al: An updated
evolutionary study of the nuclear receptor protein family. World
Acad Sci. J 3:2021.
|
|
12
|
Korostensky C and Gonnet G: Near optimal
multiple sequence alignments using a traveling salesman problem
approach. 6th International Symposium on String Processing and
Information Retrieval. 5th International Workshop on Groupware
(Cat. No. PR00268): 105-114, 1999.
|
|
13
|
Mailund T, Brodal GS, Fagerberg R,
Pedersen CN and Phillips D: Recrafting the neighbor-joining method.
BMC Bioinformatics. 7(29)2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Waterhouse AM, Procter JB, Martin DM,
Clamp M and Barton GJ: Jalview version 2-a multiple sequence
alignment editor and analysis workbench. Bioinformatics.
25:1189–1191. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mitsis T, Papageorgiou L, Efthimiadou A,
Bacopoulou F, Vlachakis D, Chrousos GP and Eliopoulos E: A
comprehensive structural and functional analysis of the ligand
binding domain of the nuclear receptor superfamily reveals highly
conserved signaling motifs and two distinct canonical forms through
evolution. World Acad Sci J. 1:264–274. 2019.
|
|
16
|
Henikoff S and Henikoff JG: . Amino acid
substitution matrices from protein blocks. Proc Natl Acad Sci USA.
89:10915–10919. 1992.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Altschul SF, Gish W, Miller W, Myers EW
and Lipman DJ: Basic local alignment search tool. J Mol Biol.
215:403–410. 1990.PubMed/NCBI View Article : Google Scholar
|
|
18
|
wwPDB consortium: Protein data bank: The
single global archive for 3D macromolecular structure data. Nucleic
Acids Res. 47:D520–D528. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Papakonstantinou E, Vlachakis D, Thireou
T, Vlachoyiannopoulos PG and Eliopoulos E: A holistic evolutionary
and 3d pharmacophore modelling study provides insights into the
metabolism, function, and substrate selectivity of the human
monocarboxylate transporter 4 (Hmct4). Int J Mol Sci.
22(2918)2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Molecular Operating Environment (MOE),
2016.0801 Chemical Computing Group ULC, 1010 Sherbooke St. West,
Suite #910, Montreal, QC, Canada, H3A 2R7, 2022.
|
|
21
|
Vangelatos I, Vlachakis D, Sophianopoulou
V and Diallinas G: Modelling and mutational evidence identify the
substrate binding site and functional elements in APC amino acid
transporters. Mol Membr Biol. 26:356–370. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Diakou KI, Mitsis T, Pierouli K,
Papakonstantinou E, Megalooikonomou V, Efthimiadou A and Vlachakis
D: Study of the langat virus RNA-dependent RNA polymerase through
homology modeling. EMBnet J. 26(e944)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Coutsias EA and Wester MJ: RMSD and
symmetry. J Comput Chem. 40:1496–1508. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gooch JW: Ramachandran Plot. In: Gooch
J.W. (eds) Encyclopedic Dictionary of Polymers. Springer, New York,
NY, 2011.
|
|
25
|
Kandil S, Biondaro S, Vlachakis D, Cummins
AC, Coluccia A, Berry C, Leyssen P, Neyts J and Brancale A:
Discovery of a novel HCV helicase inhibitor by a de novo drug
design approach. Bioorg Med Chem Lett. 19:2935–2937.
2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vlachakis D, Koumandou VL and Kossida S: A
holistic evolutionary and structural study of flaviviridae provides
insights into the function and inhibition of HCV helicase. PeerJ.
1(e74)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wadood A, Riaz M, Uddin R and Ul-Haq Z: In
silico identification and evaluation of leads for the simultaneous
inhibition of protease and helicase activities of HCV NS3/4A
protease using complex based pharmacophore mapping and virtual
screening. PLoS One. 9(2)(e89109)2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Saraste M, Sibbald PR and Wittinghofer A:
The P-loop-a common motif in ATP- and GTP-binding proteins. Trends
Biochem Sci. 15:430–434. 1990.PubMed/NCBI View Article : Google Scholar
|
|
29
|
delToro D, Ortiz D, Ordyan M, Sippy J, Oh
CS, Keller N, Feiss M, Catalano CE and Smith DE: Walker-A motif
acts to coordinate ATP hydrolysis with motor output in viral DNA
packaging. J Mol Biol. 428:2709–2729. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ruff M, Krishnaswamy S, Boeglin M,
Poterszman A, Mitschler A, Podjarny A, Rees B, Thierry JC and Moras
D: Class II aminoacyl transfer Rna synthetases: Crystal structure
of yeast aspartyl-trna synthetase complexed with trna(Asp).
Science. 252:1682–1689. 1991.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Utama A, Shimizu H, Hasebe F, Morita K,
Igarashi A, Shoji I, Matsuura Y, Hatsu M, Takamizawa K, Hagiwara A
and Miyamura T: Role of the DExH Motif of the Japanese encephalitis
virus and hepatitis C virus NS3 proteins in the ATPase and RNA
helicase activities. Virology. 273:316–324. 2000.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lee T and Pelletier J: The biology of DHX9
and its potential as a therapeutic target. Oncotarget.
7:42716–42739. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Luo D, Xu T, Watson RP, Scherer-Becker D,
Sampath A, Jahnke W, Yeong SS, Wang CH, Lim SP, Strongin A, et al:
Insights into RNA unwinding and ATP hydrolysis by the flavivirus
NS3 protein. EMBO J. 27:3209–3219. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Hollingsworth SA and Karplus PA: A fresh
look at the Ramachandran plot and the occurrence of standard
structures in proteins. BioMolecular Concepts. 1:271–283.
2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rathi PC, Ludlow RF and Verdonk ML:
Practical high-quality electrostatic potential surfaces for drug
discovery using a graph-convolutional deep neural network. J Med
Chem. 63:8778–8790. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Papageorgiou L, Vlachakis C, Dragoumani K,
Raftopoulou S, Brouzas D, Nicolaides NC, Chrousos GP, Charmandari
E, Megalooikonomou V and Vlachakis D: HCV genetics and genotypes
dictate future antiviral strategies. J Mol Biochem. 6:33–40.
2017.PubMed/NCBI
|
|
37
|
Li K, Frankowski KJ, Belon CA,
Neuenswander B, Ndjomou J, Hanson AM, Shanahan MA, Schoenen FJ,
Blagg BS, Aubé J and Frick DN: Optimization of potent hepatitis C
virus NS3 helicase inhibitors isolated from the yellow dyes
thioflavine S and primuline. J Med Chem. 55:3319–3330.
2012.PubMed/NCBI View Article : Google Scholar
|