Introduction
Ankylosing spondylitis (AS) is a progressive and
chronic inflammatory disease, which primarily involves the
sacroiliac joints and spine, though peripheral joints, including
the shoulders, hips, ribs, heels and small joints of the hands and
feet, may also be affected (1,2). The most
prevalent early symptoms are pain and stiffness in the affected
joint, which is most severe at rest, while improving during
activity (3). AS typically affects
younger individuals, developing between the ages of 15 and 30.
Occasionally, the disease affects multiple members of a family
(4).
Class I human leukocyte antigen (HLA)-B*27 has been
associated with AS in numerous studies (2). This antigen is present in over 95% of
patients with AS in the Caucasian population (5–7). In the
coding sequence for HLA-B*27 there is genetic polymorphism. In
total, 105 polymorphism subtypes exist, namely HLA-B*2701 to
HLA-B*27106, encoded by 132 alleles (8). These alleles are discriminated by
changes primarily in exons 2 and 3, which encode the α1 and α2
chains of the B27 molecule, respectively (9). HLA-B*27 typing has been proposed as a
useful test for AS and related spondyloarthropathies (SpA), and
thus the availability of a reliable and accurate detection method
is important (3). Although the result
of HLA-B*27 typing is unable confirm or exclude the diagnosis of
this disease (10), different
subtypes may be associated with different ethnic groups, clinical
manifestation, age of onset and prognosis (11).
The microlymphocytotoxicity test (MLCT) (12) and flow cytometry (13) are commonly used techniques for routine
typing of HLA-B*27. These tests rely on the detection of cell
surface antigens with antibodies. Major drawbacks of MLCT include
the requirement for viable cells, cross-reactivity of antigens, the
requirement for a single individual to provide consistent and
accurate results, and unavailability of B*27 specific antisera
covering all HLA-B*27 alleles (14).
Flow cytometry is often used due to its efficiency as an
alternative HLA-B*27 detection technique; however, cross-reactivity
with other HLA-B molecules including B7 is a major limitation
(15,16). Furthermore, it requires expensive
reagents, complex equipment and specifically trained personnel. As
a result, many laboratories have now adopted replacement molecular
methods.
The two main molecular methods most frequently
adapted for histocompatibility typing are polymerase chain reaction
with sequence specific primers (PCR-SSP) or with sequence specific
oligonucleotide probes (PCR-SSO) (14). In particular, the PCR-SSP technique
relies on the recognition of HLA-B*27 specific DNA sequences, and
thus is an ideal test for HLA-B*27. Previous results have concluded
that the outcomes obtained by PCR-SSP are more accurate compared
with the conventional serological approach (15).
PCR-SSP amplification of HLA-B*27 loci is
established as a precise and accurate method. It is based on the
basic PCR principle that under controlled conditions, primers with
complementary sequences to target sequences result in amplified
products, while non-complementary primers do not initiate
amplification (17). Nevertheless, an
internal control primer pair, which amplifies a conserved region,
is included in each PCR reaction mix in addition to
sequence-specific primers, to indicate the integrity of the PCR
reaction (18).
The present study focused on optimization and
evaluation of in-house sequence-specific PCR for HLA-B*27 alleles.
It differentiated 29 commonly occurring and disease-associated
HLA-B*27 alleles (B*2701-B*2721 and B*2723-B*2730) to the 4-digit
level (19). The determination of
associations between specific HLA-B*27 alleles with AS may aid to
identify individuals who are likely to develop the disease, and if
the population frequency of disease-associated alleles is low, then
the diagnostic accuracy of the test will be increased. Furthermore,
the identification of individuals at risk may aid to adapt
preventive strategies.
Materials and methods
Samples
A total of 39 individual HLA-B*27-positive
specimens, determined by flow cytometry (20), were obtained from patients (mean age,
31.8±8.6 years) who were symptomatic for SpA and referred to the
Armed Forces Institute of Pathology (AFIP, Rawalpindi, Pakistan)
for HLA-B*27 testing between April and December 2016. Individuals
presenting with a history of trauma and/or mechanical back pain
were excluded. Blood specimens from 10 HLA-B*27-negative transplant
donors (mean age, 18.9±16.3 years) were included as negative
controls following HLA typing using a low-resolution HLA-B typing
set consisting of 96 primer mixtures (micro SSP 1L96 wells tray;
One Lamda; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Table I lists the HLA-B specificities
of the 10 negative controls tested. The institutional review board
of AFIP approved the study and written informed consent from all
patients was obtained.
 | Table I.HLA-B specificities of the 10
HLA-B*27-negative samples tested. |
Table I.
HLA-B specificities of the 10
HLA-B*27-negative samples tested.
| HLA-B
specificity | Total samples, n |
|---|
| B*13 | 1 |
| B*14 | 1 |
| B*15 | 2 |
| B*35 | 4 |
| B*40 | 3 |
| B*44 | 2 |
| B*51 | 4 |
| B*55 | 2 |
| B*58 | 1 |
In-house PCR-SSP method
Chemicals and reagents used for HLA-B*27
amplifications. Polymerase chain reaction buffer [10X with
(NH4)2SO4; Thermo Fisher
Scientific, Waltham, MA, USA], a customized HLA-B*27 allele primer
set of 35 (Alpha DNA, Montreal, QC, Canada) (19), dNTPs, MgCl2 (both from
Thermo Fisher Scientific), HLA-DRB1 locus-specific primers
(21) as internal control primer
(Alpha DNA), Taq DNA polymerase (NovaTaq™ DNA Polymerase; EMD
Millipore, Billerica, MA, USA), a Gentra Puregene Blood kit
(Qiagen, Inc., Valencia, CA, USA), dimethyl sulfoxide (DMSO;
Scharlab S.L., Barcelona, Spain) Tris base (Ultra-Pure Tris buffer;
Invitrogen; Thermo Fisher Scientific), EDTA, boric acid (both from
Merck KGaA, Damstadt Germany), agarose (OmniPur; EMD Millipore),
bromophenol blue (Merck KGaA), gel loading dye [20% (w/v) sucrose,
0.25% (w/v) bromophenol blue], ethidium bromide (Invitrogen; Thermo
Fisher Scientific) and Mili-Q water (EvoQua, Pittsburgh, PA, USA)
were used in PCR-SSP.
DNA extraction for the amplification
of SSP
Venous blood samples (3 ml) were collected in
EDTA-potassium tubes from each patient. DNA was extracted using the
Gentra Puregene Blood kit, according to the manufacturer's
instructions. The isolated DNA was washed in ethanol and the final
concentration was adjusted to 100 ng/µl. Different concentrations
of DNA (0.1–100 ng/µl) were initially tested to obtain clear bands
for HLA-B*27 and to minimize nonspecific amplification. DNA purity
ratio was maintained between 1.7 and 2.0, determined at 260 and 280
nm using an Epoch microplate spectrophotometer (BioTek Instruments,
Inc., Winooski, VT, USA). DNA was stored at −20°C until
analysis.
PCR master mix preparation
The final optimized reaction mix was composed of the
following: DNA Taq polymerase (5 U/µl), 0.03 U/17 µl SSP reaction;
dNTPs (final concentration of each dNTP, 0.172 mM); PCR buffer, 17
mM; Tris-HCL, 64 mM, pH 8.8; MgCl2, 1.7 mM; DMSO, 2%
(v/v); internal control primers (forward and reverse), 0.23 µM
each; and Mili-Q water, 208 µl. This reaction mixture was equally
divided into 29 reaction tubes and one negative control per sample
(15 µl each).
Amplification control primers
The control primer pair included in master mix that
amplified the third intron of the DRB1 gene (21) had the following sequences: HLA-DRB1
forward, 5′-TGCCAAGTGGAGCACCCAA-3′ and reverse,
5′-GCATCTTGCTCTGTGCAGAT-3′. These primers matched conserved
sequences and thus functioned as an internal positive amplification
control.
Allele specific primers
A total of 35 sequence-specific primers for HLA-B*27
alleles (19) were purchased (Alpha
DNA) as lyophilized, salt-purified oligonucleotides. Following
reconstitution in Mili Q water, 100 µM stock solution was made,
followed by a working dilution of 3 mM. Sequences, annealing
position and specificities are listed in Table II. The primers were added to the PCR
reaction tubes (29 tubes/sample) at a final concentration of 0.23
µM. Thus, 29 SSP mixtures were made to assign 29 HLA-B*27 alleles
at the 4-digit level (B*2701-B*2721 and B*2723-B*2730).
| Table II.Human leukocyte antigen B*27 allele
primer sequences and specificities. |
Table II.
Human leukocyte antigen B*27 allele
primer sequences and specificities.
| Forward primer | Reverse primer |
|
|
|---|
|
|
|
|
|---|
| No. | Position | Sequence, 5–3 | No. | Position | Sequence, 5–3 | Amplicon size,
bp | B*27 alleles
amplified |
|---|
| 01 | 297–314 |
AGAGAACCTGCGCACCGC | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 380 | 01 |
| 02 | 149–167 |
GCTACGTGGACGACACGCT | 18 | 311–330 |
GTTGTAGTAGCGGAGCGCGA | 185 | 02, 30 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 390 | 03, 05, 09, 10, 13,
14, 16, 17, 19, 28 |
| 04 | 284–302 |
CACAGACTGACCGAGAGAG | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 390 | 04, 08, 12, 15, 18,
23, 25, 26 |
| 05 | 230–248 |
GCAGGAGGGGCCGGAGTT | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 445 | 17 |
| 06 | 254–272 |
ACCGGGAGACACAGATCTC | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 420 | 18, 29 |
| 07 | 261–282 |
ACACAGATCTACAAGACCAAC | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 410 | 12, 16, 18, 23,
29 |
| 08 | 294–311 |
CCGAGAGAGCCTGCGGAA | 17 | 411–428 |
TCGTAGGCGTCCTGGTGG | 380 | 08, 12, 18, 26 |
| 09 | 254–272 |
ACCGGGAGACACAGATCTG | 19 | 354–371 |
CCATACATCGTCTGCCAA | 365 | 14 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 20 | 363–381 |
CACGTCGCAGCCGTACATG | 340 | 07 |
| 10 | 262–283 |
CACAGATCTGCAAGGCCAAGG | 21 | 412–430 |
CGTCGTAGGCGTACTGGTC | 410 | 06, 21 |
| 11 | 262–282 |
CACAGATCTGCAAGGCCAAG | 21 | 412–430 |
CGTCGTAGGCGTACTGGTC | 410 | 06, 21 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 22 | 369–385 |
GCCCCACGTCGCAGCCG | 350 | 07, 19 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 23 | 418–434 |
CTTGCCGTCGTAGGCGTG | 395 | 09 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 24 | 418–434 |
CTTGCCGTCGTAGGCGTC | 395 | 03, 05, 10, 13, 14,
16, 17, 19, 28, 29 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 25 | 527–544 |
CTCTCAGCTGCTCCGCCT | 505 | 10 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 26 | 418–435 |
GTTTGCCGTCGTAGGCGTA | 395 | 07, 27 |
| 10 | 262–283 |
CACAGATCTGCAAGGCCAAGG | 20 | 363–381 |
CACGTCGCAGCCGTACATG | 360 | 07, 11, 24 |
| 12 | 4–14 |
CGAGATGCGGGTCACGGC | 27 | 97–114 |
CCGGGACACGGAGGTGTG | 255 | 01–12, 14–21,
23–30 |
| 13 | 4–14 |
CGAGATGCGGGTCACGGA | 27 | 97–114 |
CCGGGACACGGAGGTGTG | 255 | 13 |
| 14 | 83–97 |
CCACTCCATGAGGTATTTCC | 28 | 247–265 |
GTGTCTCCCGGTCCCAATG | 190 | 03 |
| 14 | 83–97 |
CCACTCCATGAGGTATTTCC | 29 | 247–265 |
GTGTCTCCCGGTCCCAATA | 190 | 01, 02, 04, 18–21,
24–30 |
| 02 | 149–167 |
GCTACGTGGACGACACGCT | 25 | 527–544 |
CTCTCAGCTGCTCCGCCT | 640 | 04, 06, 10, 15, 18,
20, 21, 24, 25 |
| 02 | 149–167 |
GCTACGTGGACGACACGCT | 30,31 | 280–298 |
CTCGGTCAGTCTGTGCCTT | 155 | 01–11, 13–15,
17, |
|
|
|
|
| 280–299 |
TCTCGGTAAGTCTGTGCCTT |
| 19–21, 24, 25, 27,
28, 30 |
| 02 | 149–167 |
GCTACGTGGACGACACGCT | 32 | 559–576 |
GAGCCACTCCACGCACGT | 675 | 15, 28 |
| 16 | 344–363 |
GGTCTCACACCCTCCAGAAT | 33 | 559–576 |
GAGCCACTCCACGCACAG | 235 | 25 |
| 15 | 302–319 |
CCTGCGGACCCTGCTCC | 34 | 363–383 |
CACGTCGCAGCCGTACATC | 325 | 19, 21 |
| 03 | 284–302 |
CACAGACTGACCGAGAGGA | 32 | 559–576 |
GAGCCACTCCACGCACGT | 540 | 28 |
| 09 | 254–272 |
ACCGGGAGACACAGATCTG | 35 | 363–383 |
CACGTCGCAGCCGTACATC | 370 | 19, 21, 30 |
PCR protocol
Forward and reversed primers (1 µl each) were
dispensed in 29 mini Eppendorf tubes according to the primer mix
combinations (Table II). A total of
15 µl master mix was added to each and to the negative control
tube. PCR was performed on an Eppendorf Mastercycler Gradient
thermocycler (7 tests were also run on a MultiGene OptiMax Labnet
International Thermal Cycler) at an initial denaturation
temperature of 94°C for 5 min, followed by 30 cycles of
amplification (30 sec at 95°C, 60 sec at 64°C and 72°C for 60 sec)
and a final elongation step at 72°C for 7 min, with storage at 4°C.
A total of 10 random samples were repeated.
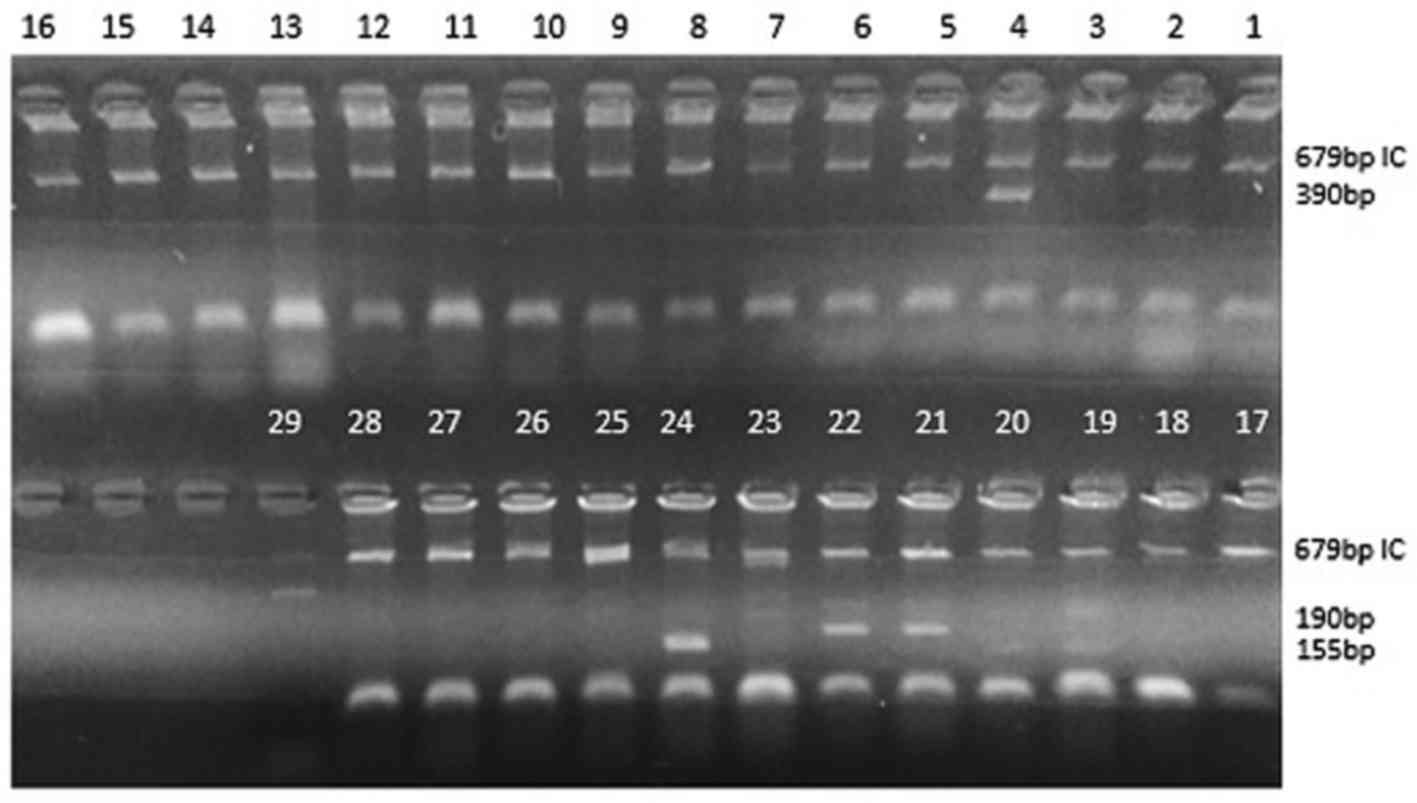
Visualization of amplifications by
agarose gel electrophoresis
Agarose gel 2% (w/v) in 0.1X Tris/borate/EDTA (TBE)
buffer was prepared. The agarose was dissolved by boiling, then
cooled to 60°C. Ethidium bromide (0.5 µg/ml gel solution) was then
added. A 4–5-mm thick gel with 3-mm wide slots was cast and set for
10–20 min. Following the addition of 1 µl loading dye to each PCR
tube, the products were loaded on the gel. The gel was run in 0.1X
TBE buffer for 35 min at 4 V/cm. Finally, the gel was examined
under ultra-violet illumination and the results were documented
(Fig. 1).
Analytical specificity of the in-house
method
The specificity of the in-house method was ensured
by the selection of the primers, as well as by concurrent use of
the micro SSP 1L96 wells typing tray, used to differentiate B27
from all other HLA-B alleles.
Results
HLA-B*27 typing
A total of 189 suspected cases of SpA were referred
to AFIP in 2016 for HLA-B*27 typing by flow cytometry, and 45 were
identified as HLA-B*27-positive (23.8%). In the present study, a
total of 49 specimens with complete medical records were assessed:
39 HLA-B*27-positive specimens determined by flow cytometry and 10
negative donor specimens that were HLA-B*27-negative, determined by
PCR-SSP.
In house PCR-SSP
Of the 39 positive samples, 31 samples exhibited
positive HLA-B*27 allele results with positive internal control,
listed in Table III (including 22
confirmed SpA cases, of which 20 were AS, with diagnosis confirmed
on positive HLA-B*27 typing), while the remaining 8 failed (low DNA
concentration) or became invalid (no internal control positive) due
to sub optimized conditions. The DRB1 internal control (product
size 796 bp), detected in 31 samples, confirmed the correct PCR
conditions and the presence of sufficient template DNA in these
amplifications. The 10 transplant donors who were HLA-B*27-negative
according to the low-resolution PCR-SSP kit were also detected as
negative by in-house PCR-SSP. Thus, overall sensitivity and
specificity were 100%, and there were no false positive or false
negative results (Table IV). Similar
results were obtained for 7 samples, which were run on two
distinctive thermal cyclers (Eppendorf Master cycler gradient and
MultiGene OptiMax Labnet International). In addition, 10 random
samples were tested twice for HLA-B*27 using the in-house PCR-SSP
test and demonstrated reproducible results.
| Table III.Positive detection of human leukocyte
antigen B*27 by in-house polymerase chain reaction with
sequence-specific primers. |
Table III.
Positive detection of human leukocyte
antigen B*27 by in-house polymerase chain reaction with
sequence-specific primers.
| SpA patient ID | Possible B*27
alleles |
|---|
| 1 | 04, 08, 12, 15, 16,
18, 19, 21, 23, 25, 26, 29 |
| 2 | 19, 21 |
| 3 | 03, 05, 10, 13, 14,
16, 17, 19, 28, 29 |
| 4 | 01, 02, 04, 18–21,
24–30 |
| 5 | 01, 02, 04, 18–21,
24–30 |
| 6 | 12, 16, 18, 19, 21,
23, 29, 30 |
| 7 | 19 |
| 8 | 02, 12, 16, 18, 19,
21, 23, 29, 30 |
| 9 | 30 |
| 10 | 07, 19 |
| 11 | 06, 07, 11, 21,
24 |
| 12 | 07 |
| 13 | 06, 21 |
| 14 | 06, 21, 25 |
| 15 | 03 |
| 16 | 07 |
| 17 | 21 |
| 18 | 07, 10, 19 |
| 19 | 10 |
| 20 | 10 |
| 21 | 07 |
| 22 | 03, 05, 10, 13, 14,
16, 17, 19, 28, 29 |
|
| Suspected SpA
patient ID | Possible B*27
alleles |
|
| 23 | 01, 03, 05, 06, 07,
10, 13, 14, 16, 17, 19, 21, 28, 29 |
| 24 | 03, 05, 07, 10, 11,
13, 14, 16, 17, 19, 24, 28 29 |
| 25 | 07, 19, 21 |
| 26 | 14, 19, 21, 30 |
| 27 | 19, 21, 30 |
| 28 | 07, 19 |
| 29 | 07, 11, 24 |
| 30 | 07, 19, 21 |
| 31 | 01–11, 13–15, 17,
19–21, 24, 25, 27, 28, 30 |
| Table IV.Comparison of flow cytometry and
PCR-SSP. |
Table IV.
Comparison of flow cytometry and
PCR-SSP.
| In-house
PCR-SSP | Flow cytometry
Positive | Low-resolution
PCR-SSP (kit) Negative | Total |
|---|
| Positive | 31 | 0 | 31 |
| Negative | 0 | 10 | 10 |
| Total | 31 | 10 | 41 |
Analytical sensitivity
Analytical sensitivity of the PCR-SSP technique was
tested with different concentrations of DNA ranging from 0.1 to 100
ng/µl. Positive bands of the control and B*27-specific primers were
most obviously detected at concentrations ranging from 50 to 92
ng/µl.
Discussion
The association between AS and HLA-B*27 alleles
depends on ethnicity and geographical location. HLA-B*27 is more
frequent in Punjabi and Urdu-speaking communities in Pakistan
(22). Certain alleles exhibit an
association with increased susceptibility to AS, including B*2702,
B*2704, B*2705, B*2707 and B*2708 (23), while others may confer protection,
including B*2706 and B*2709 (24).
Thus, subtype identification may be helpful for early diagnosis and
improved prognosis of AS.
A PCR detection method for HLA-class I and II genes
has been used by us for tissue typing of transplant patients, and
was used as a starting point in the present study. PCR-SSP for
HLA-B*27 detection is a method based on the use of specific primers
complementary to unique HLA-B*27 gene sequences. In the present
study, an in-house PCR-SSP test was developed which amplified
common HLA-B*27 alleles (2701–2721 and 2723–2730) (19).
The specificity of PCR is influenced by numerous
factors, including the sequence of the primer (GC content), ratio
of primer, buffer, Mg2+ ion concentration and Taq
polymerase concentration (25,26). All
conditions were optimized in the present study for all primer pairs
in succession. This optimization resulted in a slightly different
protocol from tissue typing; the primer concentration was lower and
the optimal annealing temperature was higher. However, the results
may contradict as when primer concentration is lower, more cycles
and higher annealing temperature are required (26). The lower primer concentration was
important to prevent primer dimerization. Initially in preliminary
assays, 0.47 µmol/µl primer was used in each reaction, which was
identified to create primer-dimers (data not shown). Reducing
primer concentration to 0.23 µmol/μl minimized the presence of
these dimers. Another important variable in PCR is the
MgCl2 concentration; an optimal concentration of 1.7 mM
was achieved in the present study by titration. Furthermore, the
annealing temperature required optimization, which was increased to
64°C, resulting in a higher specificity of synthesis of HLA-B*27
products. The DRB1 product also exhibited improved intensity with
higher annealing temperature. Therefore, an optimized annealing
temperature was important to avoid false positive bands, which may
otherwise lead to misdiagnosis of patients. DMSO may have also
improved the PCR reaction, by decreasing inter- and intra-strand
reannealing (27–29). Additionally, precautionary measures
were taken to prevent contamination of samples using dedicated
equipment and reagents.
The analytical sensitivity of the PCR-SSP technique
was tested with different concentrations of DNA ranging from 0.1 to
100 ng/µl. Positive bands of the control and B*27-specific primers
were most obviously detected at concentrations ranging from 50 to
92 ng/µl. High analytical sensitivity of in-house PCR-SSP has
advantages over other methods, as it requires lower quantity blood
samples compared with MLCT, and older samples may be used compared
with flow cytometry, which requires fresh samples for accurate
results.
The present study represents an advancement in
biomedical science in Pakistan, as to the best of our knowledge, it
is a pioneer attempt in this region, although this work has been
conducted in other countries. Immunological research is
comparatively novel in Pakistan and there are few available
molecular immunology laboratories. The association of different
HLAs with autoimmune diseases is now an active area of research in
the region.
The cost of the present in-house, optimized PCR SSP
technique was relatively low; the technique was ~2 times less
costly than that of imported commercial PCR-SSP test kits provided
by certain laboratories. Thus, in developing countries including
Pakistan, in-house protocols provide a more economical alternative.
Nevertheless, applications of the present study may lead to
improved understanding and treatment of general SpA diseases.
Furthermore, it may form the basis for the future studies on the
association of HLA-B*27 alleles with AS and related
arthropathies.
In conclusion, in-house PCR-SSP should be considered
a standard method for the detection of HLA-B*27 alleles. It is a
relatively accurate method that bypasses the typical problems
associated with MLCT and flow cytometry, and may improve the
diagnosis of AS at the molecular level. Furthermore, although the
technique is labour intensive, it is markedly less costly than
imported commercial PCR-SSP test kits.
References
|
1
|
Ahsan T, Erum U, Jabeen R and Khowaja D:
Ankylosing Spondylitis: A rheumatology clinic experience. Pak J Med
Sci. 32:365–368. 2016.PubMed/NCBI
|
|
2
|
Hospital TO: The pathogenesis of
ankylosing spondylitis. Neurosurg Focus. 24:1–10. 2008. View Article : Google Scholar
|
|
3
|
Raychaudhuri SP and Deodhar A: The
classification and diagnostic criteria of ankylosing spondylitis. J
Autoimmun 48–49. 1–133. 2014.
|
|
4
|
Reveille JD: Major histocompatibility
genes and ankylosing spondylitis. Best Pract Res Clin Rheumatol.
20:601–609. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brown MA: Breakthroughs in genetic studies
of ankylosing spondylitis. Rheumatology (Oxford). 47:132–137. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheehan NJ and Frcp M: The ramifications
of HLA-B27. J R Soc Med. 97:10–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reveille JD, Ball EJ and Khan MA: HLA-B27
and genetic predisposing factors in spondyloarthropathies. Curr
Opin Rheumatol. 13:265–272. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khan MA: Polymorphism of HLA-B27: 105
subtypes currently known. Curr Rheumatol Rep. 15:1–6. 2013.
View Article : Google Scholar
|
|
9
|
Tipu HN: Journal of Genetic Disorders and
Genetic Reports Mini Review: HLA B27 and its immunogenetics in
ankylosing spondylitis. J Genet Disor Genet Rep. 2:12013.
|
|
10
|
Khan MA and Khan MK: Diagnostic value of
HLA-B27 testing ankylosing spondylitis and Reiter's syndrome. Ann
Intern Med. 96:70–76. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mou Y, Zhang P, Li Q, Lin Z, Liao Z, Wei Q
and Gu J: Clinical features in Juvenile-Onset ankylosing
spondylitis patients carrying different B27 subtypes. Biomed Res
Int. 2015:5948782015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dunky A, Neumüller J, Hübner C, Fischer
GF, Bayer PM, Wagner E, Schwartz DW and Mayr WR: HLA-B27
determination using serological methods. A comparison of enzyme
immunoassay and a microlymphocytotoxic test with flow cytometry and
a molecular biological assay. Rheumatol Int. 16:95–100. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Skalska U, Kozakiewicz A, Maśliński W and
Jurkowska M: HLA-B27 detection - comparison of genetic
sequence-based method and flow cytometry assay. Reumatologia.
53:74–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Middelton D: HLA typing from serology to
sequencing Era. Iran J Allergy Asthma Immunol. 4:53–66.
2005.PubMed/NCBI
|
|
15
|
Seipp MT, Erali M, Wies RL and Wittwer C:
HLA-B27 typing: Evaluation of an allele-specific PCR melting assay
and two flow cytometric antigen assays. Cytometry B Clin Cytom.
63:10–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cianga P, Zlei M, Rezus E and Cianga C:
The flow cytometric labeling pattern in HLA-B27 detection may
suggest cross reactivities. Rev Romana Med Lab. 19:185–191.
2011.
|
|
17
|
Chatterjee A, Sinha SK and Mukherjee G: A
comprehensive review on HLA and its detections by polymerase chain
reaction technique. Int J Pharm Res Sch. 3:340–346. 2014.
|
|
18
|
Shyamala V and Ferro-Luzzi Ames G: Single
specific primer-polymerase chain reaction (SSP-PCR) and genome
walking. Methods Mol Biol. 15:339–348. 1993.PubMed/NCBI
|
|
19
|
Downing J, Coates E, Street J, Hammond L,
Rees TJ, Pepperall J and Darke C: A high-resolution polymerase
chain reaction-sequence-specific primer HLA-B*27 typing set and its
application in routine HLA-B27 testing. Genet Test. 10:98–103.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hücre A, Hla B, Testinin T and Yorumu K:
Accurate and simple interpretation of HLA B27 screening by flow
cytometry. Turk J Immunol. 4:1–6. 2016. View Article : Google Scholar
|
|
21
|
Olerup O and Zetterquist H: HLA-DR typing
by PCR amplification with sequence-specific primers (PCR-SSP) in 2
hours: An alternative to serological DR typing in clinical practice
including donor-recipient matching in cadaveric transplantation.
Tissue Antigens. 39:225–235. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zafar N, Khan S, Qadir A, Raza Y, Naqvi A
and Rizvi A: Hla frequencies in Pakistani population. JPMA.
46:12–13. 1996.
|
|
23
|
Mou Y, Wu Z, Gu J, Liao Z, Lin Z, Wei Q,
Huang J and Li Q: HLA-B27 polymorphism in patients with juvenile
and adult-onset ankylosing spondylitis in Southern China. Tissue
Antigens. 75:56–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng X, Mei Y, Ji X, Xue Q and Chen D:
Molecular mechanism of the susceptibility difference between
HLA-B*27:02/04/05 and HLA-B*27:06/09 to ankylosing spondylitis:
substitution analysis, MD simulation, QSAR modelling, and in vitro
assay. SAR QSAR Environ Res. 27:409–425. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sharma N, Singh P, Nautiyal SC, Shuaib M,
Rawat P and Kaur G: Rapid detection of HLA-B27 sequence specific
alleles by in-house optimized molecular assay for the detection of
ankylosing spondylitis. J Biomed Pharm Res. 2:175–180. 2013.
|
|
26
|
Yang R, Zhang JH and Yuan GY:
Establishment of an optimized PCR method using sequence-specific
primers for screening multiple gene polymorphisms simultaneously.
Mol Med Rep. 7:201–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hardjasa A, Ling M, Ma K and Yu H:
Investigating the effects of DMSO on PCR fidelity using a
restriction digest-based method. J Exp Microbiol Immunol.
14:161–164. 2010.
|
|
28
|
Hung T, Mak K and Fong K: A specificity
enhancer for polymerase chain reaction. Nucleic Acids Res.
18:49531990. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ralser M, Querfurth R, Warnatz HJ, Lehrach
H, Yaspo ML and Krobitsch S: An efficient and economic enhancer mix
for PCR. Biochem Biophys Res Commun. 347:747–751. 2006. View Article : Google Scholar : PubMed/NCBI
|