Introduction
Acute myeloid leukemia (AML) is a highly
heterogeneous hematologic malignancy with great variability in
biological, phenotypic and prognostic behaviors and strikingly
diverse outcomes to standard therapy (1,2).
Despite significant advances in the treatment of patients with AML,
the prognosis of many patients is still uncertain and optimal
post-remission therapy is unclear. Thus, the elucidation of AML
survival events is important and may potentially aid in the
prognosis and treatment of patients with AML.
In recent years, a number of studies on prognostic
markers in AML have focused on non-coding RNAs (ncRNAs), which lack
protein-coding potential. The majority of these studies have
focused on miRNAs (3–6). However, there are few reports on
long non-coding RNA (lncRNA) as prognostic markers in AML. lncRNAs
are transcripts which are >200 nucleotides in length, located
within intergenic stretches or overlapping antisense transcripts of
protein-coding genes. They have emerged as important regulators of
gene expression, showing cell-specific expression patterns and
subcellular localization and are involved in many biological
functions, including cell apoptosis, proliferation and the cell
cycle (7,8). Some studies have demonstratd that
lncRNAs are associated with AML. For example, lncRNA CCD26 has been
shown to control the growth of myeloid leukemia cells through the
regulation of KIT expression (9). Thus, it is reasonable that lncRNAs
may be considered as prognostic biomarkers.
ncRNAs rarely function in isolation, but always
function together to form biological modules (10). These functional biological modules
are often considered to be prognostic biomarkers due to their
improved robustness and interpret-ability (11). A number of methods have been
developed to discover functional modules, such as weighted gene
co-expression network analysis (WGCNA). WGCNA is a systems
biology-based approach, which offers a promising technique for
detecting functional modules (12). WGCNA has been widely used to
identify functional modules that contribute to phenotypic traits in
various diseases (13–17). Compared with other techniques
based on gene expression profiling network analysis, such as
cytoscape-based approaches, WGCNA transforms gene expression
profiles into functional co-expressed gene modules, which do not
rely on prior assumptions about genes or covariates, thereby
providing insight into biological signaling networks that may be
associated with phenotypic traits of interest (18). In this study, we used WGCNA to
identify lncRNA co-expression modules.
The Cancer Genome Atlas (TCGA) stores comprehensive
datasets of multiple cancers, including clinical data and
transcriptome data of AML. The expression levels of lncRNAs and
mRNAs in AML were calculated using RNA-seq V2 dataset. There is
evidence to indicate that lncRNAs may play a functional role by
regulating gene expression, predominantly by their secondary
structures, which is difficult to decipher (19). Considering the challenges in
investigating the functional mechanisms of lncRNA modules, a
co-expression mRNA-based method was used in this study, in which
the functions of lncRNA modules were predicted according to their
co-expressed protein-coding gene (19).
In this study, to identify prognosis-related lncRNA
modules and the potential mechanisms of AML, the expression of
lncRNAs was calculated using the RNA-seq V2 dataset of TCGA and an
AML-related lncRNA co-expression network was constructed.
Subsequently, WGCNA was used to identify AML functional lncRNA
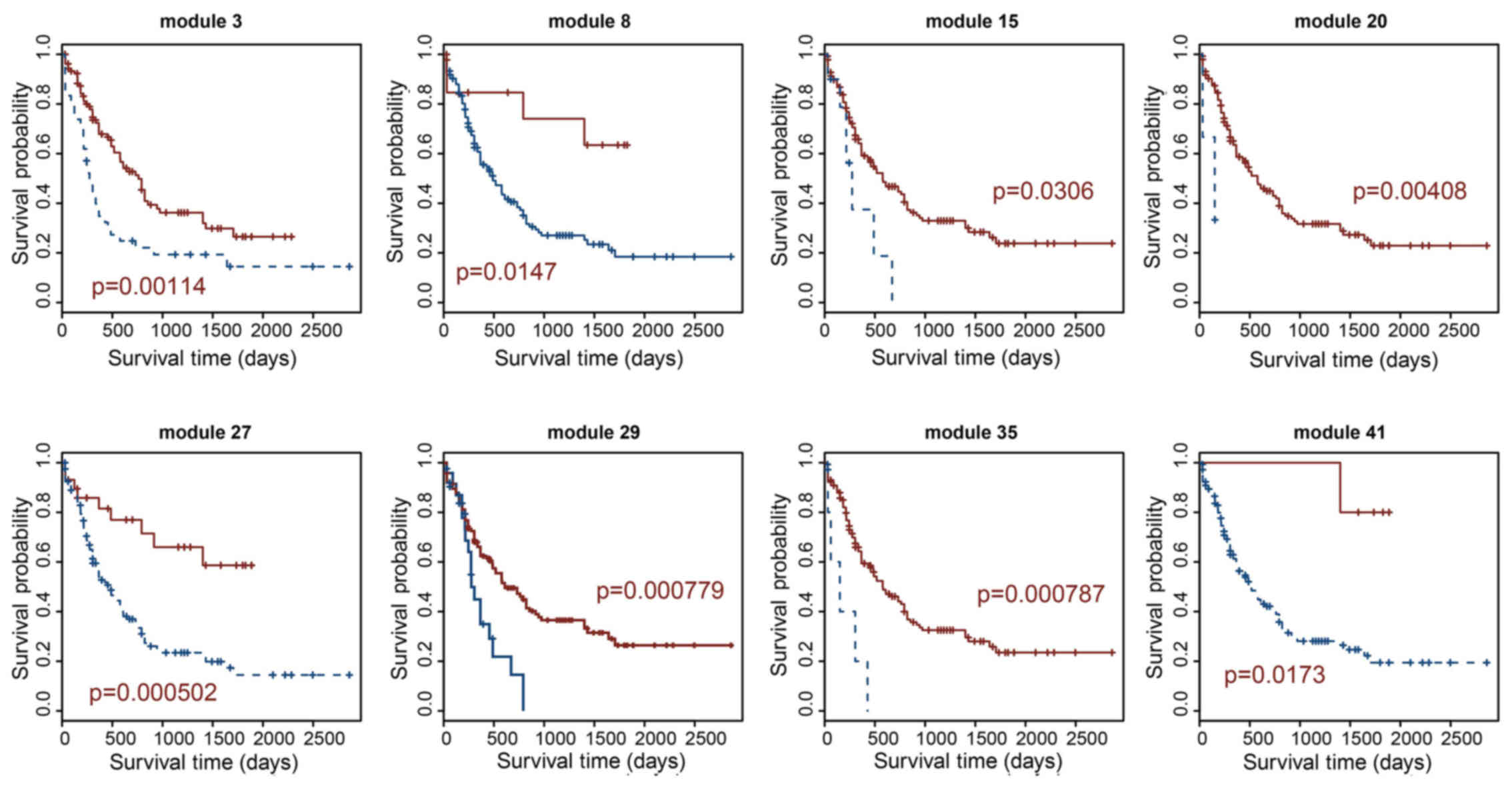
co-expression modules. Based on survival analysis, 8
prognosis-related lncRNA modules for AML were identified. Module 27
was the most significant prognosis-related lncRNA module, which
displayed the best performance in the survival prediction (log-rank
test, p=0.000502). To investigate the mechanisms of action of these
prognosis-related lncRNA modules, pathway enrichment of all
co-expressed mRNAs of lncRNA modules was implemented, and a
prognosis module-pathway network was constructed to interpret the
mechanisms of AML. The results of the present study not only
provide potential lncRNA modules as prognostic biomarkers, but also
provide further insight into the molecular mechanisms of action of
lncRNAs.
Materials and methods
Data
The RNA-seq data set of AML was downloaded from TCGA
(https://tcga-data.nci.nih.gov/). This
dataset was derived from the tissue samples of 200 adult patients
with de novo AML using RNA-seq technology. The clinical
survival data was also obtained from TCGA. Survival time was
defined as the time from tissue removal to death, loss-to-follow-up
or study conclusion. Patients who were lost to follow-up or
survival time after <20 days were deleted from the next survival
analysis. Finally, a total of 161 clinical samples remained in this
study.
Expression of lncRNAs and mRNAs in
AML
The RNA-seq V2 dataset of AML data was downloaded
from the TCGA database, with quantile-normalized and
background-corrected at level 3. The reads per kilobases per
million reads (RPKM) values of genes and lncRNAs were calculated
from exon read counts data, with RPKM = (raw read counts
×106)/(total reads x length of lncRNA/gene), in which,
the raw read counts represented all exon read counts that mapped
into a certain lncRNA/gene, and total reads were all exon read
counts that mapped into all lncRNAs/genes of one single sample.
Construction of lncRNA-lncRNA
co-expression network
The expression values of lncRNAs were obtained as
described above. Next, if the missing rate of lncRNA or mRNA
expression was >90%, the AML patients were excluded from this
study. Finally, we obtained 1,406 lncRNAs across 173 AML patients.
Pearson's correlation coefficient (PCC) and significant p-value
were calculated between the expression values of each lncRNA-lncRNA
pair across all AML samples. The lncRNA-lncRNA pairs with a p-value
<0.001 and absolute value of PCC >0.5 were used to construct
a lncRNA-lncRNA co-expression network (LLCN). Finally, the LLCN was
constructed with lncRNAs nodes, which were connected based on the
significance of co-expression between them (Fig. 2).
Identification of prognosis-related
lncRNA modules for AML
Following the construction of the LLCN, the lncRNA
co-expression modules were identified by a WGCNA R-based package,
which could extract functional modules based on pairwise correlated
expression among lncRNAs with co-regulation implications. Prior to
the identification of lncRNA co-expression modules using WGCNA,
sample clustering was first used to detect outliers. As a result,
one sample identified as an outlier was excluded from the analysis
(data not shown). In this study, we defined the prognosis-related
lncRNA modules for AML as these modules are significantly related
to the survival time of AML patients. To identify prognosis-related
lncRNA modules, the lncRNAs were extracted from each lncRNA
co-expression module as signatures to perform survival analysis.
Firstly, AML samples were divided into 2 groups based on the
expression values of signature lncRNAs for each lncRNA
co-expression module using a K-mean clustering method (20). The survival differences between 2
groups were then assessed by Kaplan-Meier estimate and compared
using a log-rank test, where a p-value <0.05 was considered to
indicate a significant result. The lncRNA co-expression module was
considered to be a prognosis-related lncRNA module.
Functional analysis of AML-related lncRNA
modules
To better illustrate the potential functional
mechanism of prognosis-related lncRNA modules of AML, the Kyoto
encyclopedia of genes and genomes (KEGG) pathway and gene ontology
(GO) functional enrichment analyses were carried out using the
database for annotation, visualization and integrated discovery
(DAVID) (21), which consists of
an integrated biological knowledge base and analytic tools aimed at
systematically extracting biological meaning from large
gene/protein lists. To do this, firstly, co-expression correlation
and significance were calculated with PCC between the expression
values of mRNA and lncRNA in prognosis-related lncRNA module across
all matched AML samples. Subsequently, for each prognosis-related
lncRNA module, the mRNA with Pearson's correlation test p-value
<0.001 and absolute value of PCC >0.5 were used for
functional enrichment analysis to investigate the mechanism of this
module in AML.
Results
In this study, we identified AML prognosis-related
lncRNA co-expression modules and interpreted their functional
mechanisms in AML. The framework of this study is shown in Fig. 1. Firstly, we obtained clinical
data and RNA-seq V2 data from TCGA. The expression values of
lncRNAs and mRNAs in all AML patients were calculated.
Subsequently, a lncRNA co-expression network was constructed by
calculating co-expression PCC. To identify prognosis-related lncRNA
modules for AML, clinical survival data were also used to identify
lncRNA modules which are significantly associated with the survival
time of AML patients. Lastly, we performed functional enrichment
analysis using mRNA with expression values correlated with lncRNAs
in each prognosis-related lncRNA module to interpret the mechanisms
of AML.
Construction of LLCN
Based on co-expression correlations identified
between each lncRNA-lncRNA pair across all AML samples with
lncRNA-lncRNA pairs with a p-value <0.001 and an absolute value
of PCC >0.5, the LLCN was constructed. The LLCN contained a
total of 1,870 lncRNAs with 83,135 edges between them (Fig. 2) (data not shown). The degree of
distribution of lncRNA nodes of LLCN followed power law
distribution with correlations of 0.910 and R2=0.877,
lncRNA RP11-492A10.1 with the highest degree.
Identification of prognosis-related
lncRNA modules for AML
To identify prognosis-related lncRNA modules for
AML, firstly, we identified functional lncRNA co-expression modules
based on LLCN using WGCNA. A total of 42 co-expression lncRNA
modules were identified, the largest module contained 427 lncRNAs
and the smallest module contained 7 lncRNAs, with an average 44.7
lncRNAs per co-expression module (data not shown). Subsequently,
for each co-expression lncRNA module, we performed survival
analysis to determine whether it is a prognosis-related lncRNA
module. In this process, AML patients were divided into 2 risk
groups according to expression values of all lncRNAs contained in
each lncRNA co-expression module in the lncRNA expression profile,
and p-value calculations were based on log-rank test (Materials and
methods). lncRNA co-expression modules with a p-value <0.05 were
considered as prognosis-related lncRNA modules. Finally, we
obtained 8 prognosis-related lncRNA modules from 42 lncRNA
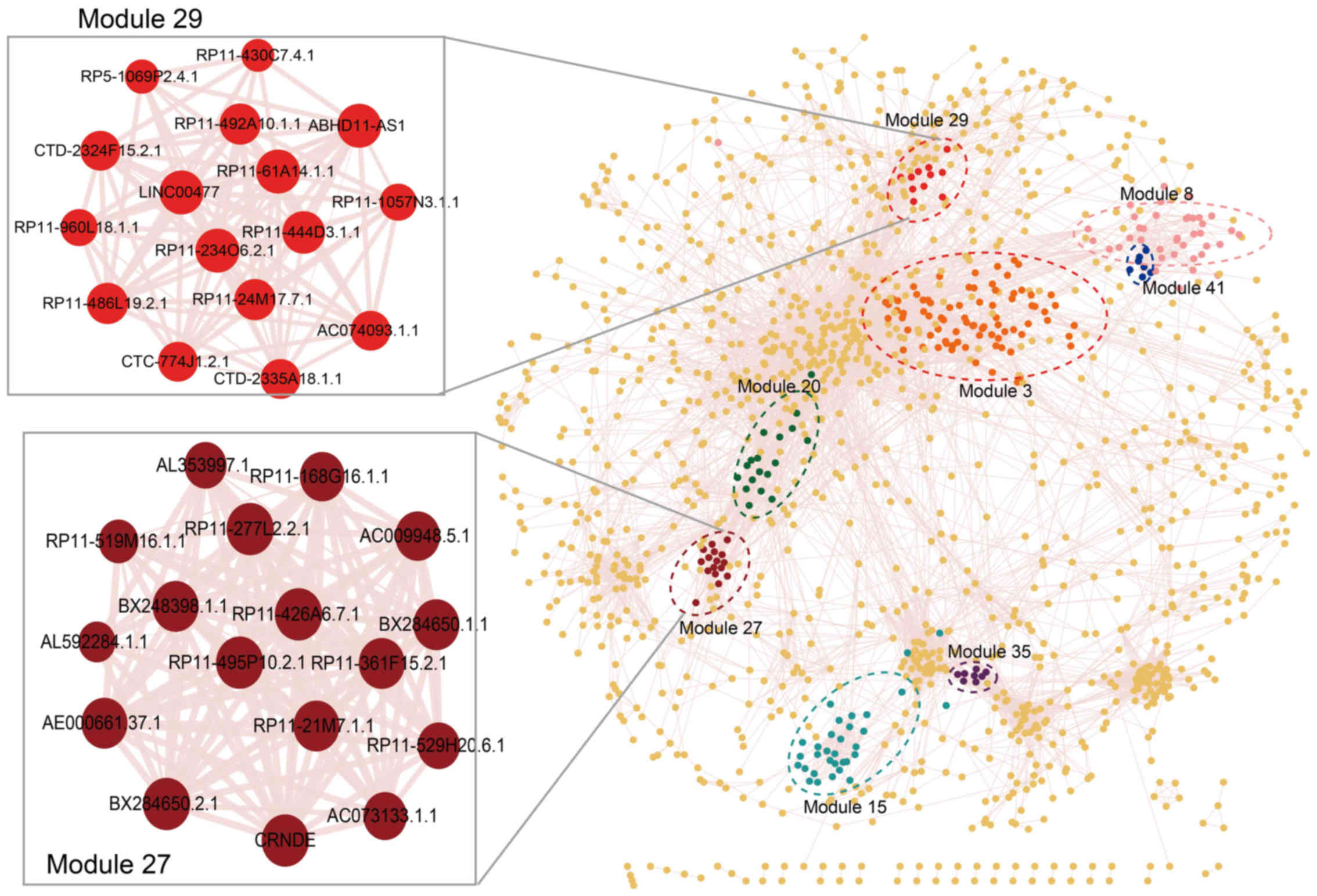
co-expression modules (Figs. 2
and 3). As shown in Table I, the largest prognosis-related
lncRNA module contained 88 lncRNAs (module 3) and the smallest
contained 7 lncRNAs (module 41). Module 27 is the most significant
prognosis-related lncRNA module, which displayed the optimal
performance in the survival prediction (log-rank test p=0.000502).
This module consisted of 17 lncRNAs; module 29 was the second most
significant prognosis-related lncRNA module (log-rank test
p=0.000779), which contained 16 lncRNAs (Fig. 2 and Table I).
 | Table IInformation of lncRNA co-expression
modules. |
Table I
Information of lncRNA co-expression
modules.
| Module name | lncRNA no. | P-value | lncRNAs |
|---|
| 27 | 17 | 0.000502 | BX284650.1.1;
BX284650.2.1; BX248398.1.1; AL592284.1.1; RP11-495P10.2.1;
RP11-277L2.2.1; RP11-21M7.1.1; AC009948.5.1; RP11-519M16.1.1;
RP11-361F15.2.1; AC073133.1.1; RP11-426A6.7.1; AE000661.37.1;
RP11-529H20.6.1; RP11-168G16.1.1; CRNDE; AL353997.1 |
| 29 | 16 | 0.000779 | RP11-430C7.4.1;
AC074093.1.1; RP11-234O6.2.1; CTD-2324F15.2.1; RP11-492A10.1.1;
ABHD11-AS1; RP11-1057N3.1.1; CTC-774J1.2.1; RP11-444D3.1.1;
LINC00477; RP11-24M17.7.1; CTD-2335A18.1.1; RP11-61A14.1.1;
RP11-960L18.1.1; RP11-486L19.2.1; RP5-1069P2.4.1 |
| 35 | 9 | 0.000787 | AC096579.13.1;
AC096579.7.1; AC096670.3.1; RP11-133O22.6.1; RP11-492E3.2.1;
SFTA1P; AL928742.12.1; RP11-731F5.2.1; AL928768.3.1 |
| 3 | 88 | 0.00114 | RP11-206L10.11.1;
RP11-181G12.2.1; RP11-576I22.2.1; RP11-145A3.1.1; AC104695.3.1;
AC013271.3.1; AC013268.5.1; RP11-1223D19.1.1; AC112229.7.1;
ENSG00000273471; RP13-1039J1.2.1; AC009299.3.1; RP11-446H18.3.1;
SMARCA5-AS1; RP11-1336O20.2.1; AC091878.1.1; ENSG00000273345;
RP1-90J20.7.1; RP11-553A21.3.1; RP11-162J8.2.1; RP11-351J23.1.1;
AC005027.4.1; RP11-875O11.2.1; RP11-177H13.2.1; CTD-3107M8.4.1;
RP11-68L18.1.1; HAS2-AS1; RP11-273G15.2.1; RP11-498P14.5.1;
RP11-308N19.4.1; ZNF883; RP11-203J24.9.1; RP11-499P20.2.1;
RP11-80K21.1.1; RP11-106M7.1.1; RP11-326C3.11.1; AC104389.28.1;
AC015691.13.1; SPON1; SHANK2-AS1; RP11-356J5.12.1; RP11-598F7.4.1;
CACNA1C-AS1; RP11-436I9.2.1; RP11-118B22.2.1; RP11-495K9.3.1;
RP11-394A14.2.1; LINC00355; MIR4500HG; RP11-44N21.4.1;
RP11-941F15.1.1; WASIR2; RP11-314O13.1.1; AC021593.1; AC105337.1;
AC127496.3; ENSG00000266149; ENSG00000265257; ENSG00000265485;
CTD-2666L21.1.1; ENSG00000267147; ENSG00000269110; AC092296.1.1;
AC092295.7.1; AC012309.5.1; ENSG00000267470; ENSG00000267640;
AC016582.2.1; ENSG00000268262; AC011497.1; ENSG00000267058;
ENSG00000267188; CEACAM20; AC011450.1; ENSG00000269959; LINC00085;
ENSG00000267827; ENSG00000267454; AC004696.1; RP4-694B14.5.1;
C20orf203; AF127936.7.1; LINC00319; LINC00313; LL22NC03-86G7.1.1;
RP4-756G23.5.1; Z83851.3.1; LL0XNC01-116E7.2.1 |
| 15 | 35 | 0.00408 | RP5-1071N3.1.1;
AC087590.3.1; RP11-81N13.1.1; RP11-553L6.2.1; RP11-18H21.1.1;
LINC00243; LINC00336; AC002480.3.1; NEFL; RP11-622O11.2.1;
RP11-234A3.1.1; RP11-229P13.19.1; RP13-25N22.1.1; RP11-730K11.1.1;
RP11-627G23.1.1; RP11-493L12.2.1; ENSG00000266923; RP11-173C20.1.1;
RP11-90M5.1.1; LINC00402; RP11-58E21.3.1; ENSG00000273065;
CTD-2506J14.1.1; RP11-23P13.6.1; ENSG00000266088; ENSG00000267128;
RP11-77C3.3.1; LINC00494; RP4-669H2.1.1; LINC00161; MIAT;
CTA-373H7.7.1; RP11-265P11.2.1; DGKK; RP3-527F8.2.1 |
| 8 | 54 | 0.0147 | TTLL10-AS1;
RP11-12L8.1.1; AC011893.3.1; AC005042.4.1; RP11-584P21.2.1;
RP11-422J15.1.1; RP11-121L11.1.1; AC034220.3.1; ENSG00000273299;
XKR5; LINC00051; FAM201A; RP11-195E11.3.1; ENSG00000268364;
RP11-154D17.1.1; RP11-492E3.1.1; RP11-144G6.12.1; ENSG00000270119;
RP11-464F9.1.1; ENSG00000271816; ENSG00000272140; ENSG00000271880;
AGAP11; ENSG00000272508; RP11-399L7.2.1; RP11-554A11.6.1;
RP11-166D19.1.1; RP11-820L6.1.1; RP11-713P17.3.1; C14orf167;
RP11-829H16.3.1; RP11-193F5.1.1; RP4-647C14.2.1; LINC00341;
RP11-37C7.1.1; RP11-358M11.2.1; RP11-809H16.2.1; ENSG00000272298;
AC091172.1; ENSG00000261924; CHST9-AS1; ENSG00000267013;
ENSG00000267629; ENSG00000267339; FXYD1; RP4-779E11.3.1;
ENSG00000270408; RP4-568F9.6.1; RP4-705O1.1.1; AL592528.1.1;
RP1-274L7.1.1; AC004383.4.1; AC004383.5.1; RP1-260J9.2.1 |
| 41 | 7 | 0.0173 | AC113607.1.1;
RP11-356I2.4.1; RP11-504G3.1.1; AP003025.2.1; MEG3; MEG8;
AL132709.7.1 |
| 20 | 22 | 0.0306 | RP11-98D18.9.1;
RP11-466F5.8.1; AC007246.3.1; AC010127.3.1; ENSG00000271270;
CTD-2049J23.2.1; RP11-497D6.4.1; AC091729.9.1; RP4-555L14.5.1;
RP11-539E17.4.1; RP11-143M1.3.1; ENSG00000271086; RP11-554F20.1.1;
LINC00202; RP11-887P2.1.1; DIO3OS; ENSG00000262188; LINC00470;
RP11-795F19.1.1; ENSG00000269696; BX322557.10.1; AP001476.2.1 |
Functional analyses of lncRNA modules in
AML
To further investigate the mechanisms of action of
prognosis-related lncRNA modules in AML, we performed functional
pathway enrichment analysis using co-expression genes of lncRNAs in
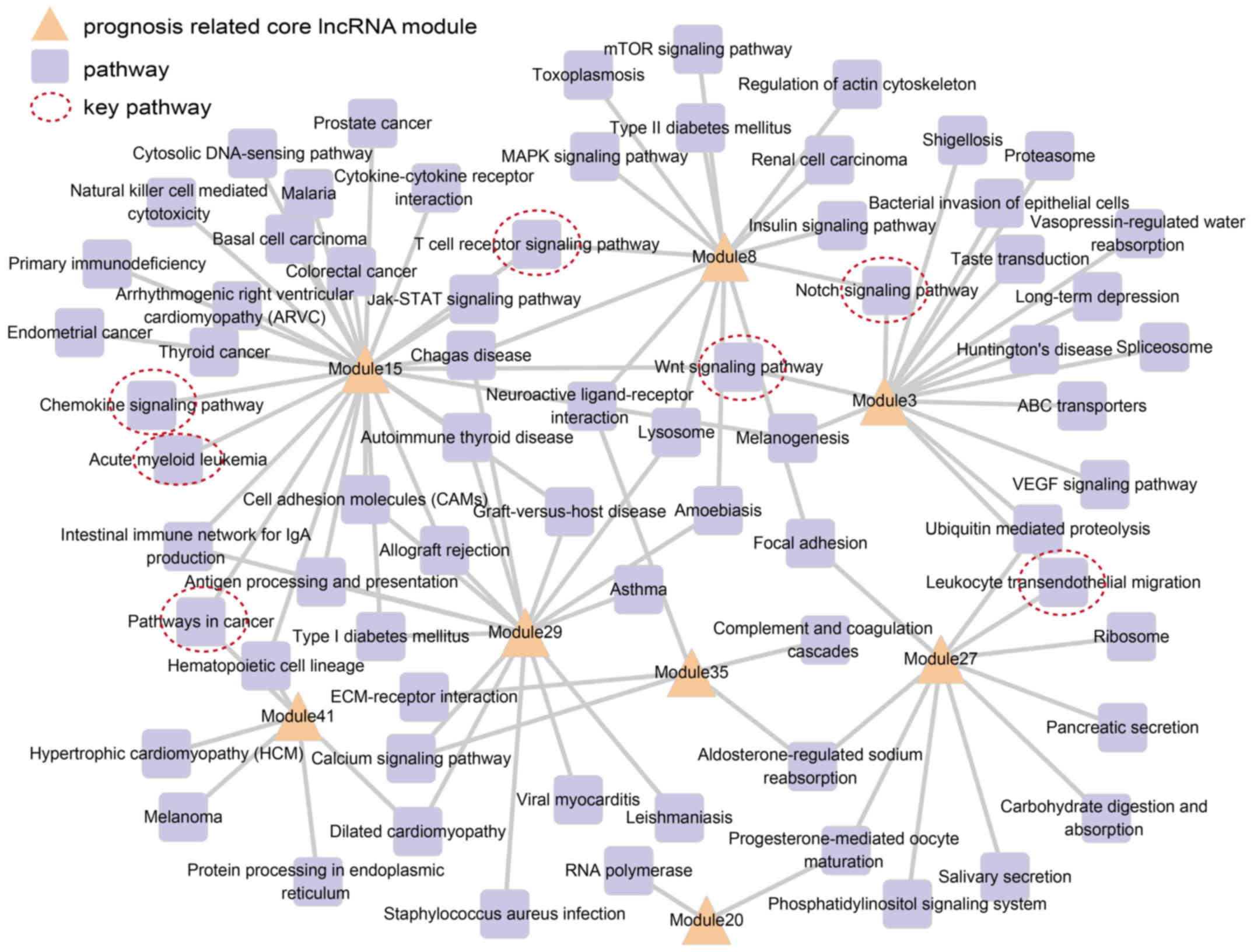
each lncRNA module. We found that 8 prognosis-related lncRNA
modules wee significantly enriched in 70 pathways, which included
certain AML-related pathways, such as the acute myeloid leukemia
pathway, chemokine signaling pathway (22), pathway in cancer, as well as
others. Subsequently, a prognosis module pathway network was
constructed based on these pathways, in which nodes represent
lncRNA modules and pathways, and edges represent significantly
enriched pathways (Fig. 4).
Fig. 4 shows all
prognosis-related lncRNA modules connected to more than one
functional pathway. Among these, modules 15, 29 and 3 were involved
in the most number of pathways, namely 27, 16 and 15, respectively.
The most significant prognosis-related module, module 27, was
involved in 10 pathways, including the leukocyte transendothelial
migration pathway (23). This
indicated that these prognosis lncRNA modules executed multiple
biological functions. We also noted that some pathways were related
with more than one lncRNA module, saling pathway (24), notch signaling pathway (25) and T cell receptor signaling
pathway (26–28).
Discussion
In the present study, the expression levels of
lncRNAs and mRNAs in AML were calculated, following which an LLCN
was constructed (Fig. 2). This
network contained 1,406 lncRNAs with 7,915 edges between them. The
most connected lncRNA was RP11-492A10.1, which is an anti-sense RNA
locating to chromosome 15. To investigate the function of
RP11-492A10.1, pathway enrichment analysis was performed on 125
protein-coding genes that co-expressed with RP11-492A10.1. The
results revealed that these genes were involved in the neuroactive
ligand-receptor interaction pathway, calcium signaling pathway, and
chemokine signaling pathway. It has been demonstrated that CXCR4
chemokine receptor signaling induces the apoptosis of in AML cells
via the regulation of the Bcl-2 family members, Bcl-xL, Noxa and
Bak (22). This suggested that
RP11-492A10.1 may be an important regulator in AML via co-expressed
genes.
To identify prognosis-related lncRNA modules for
AML, WGCNA was used to detect functional lncRNA co-expression
modules. By integrating clinical survival data, 8 prognosis-related
lncRNA modules were identified from 42 lncRNA co-expression modules
(Figs. 2 and 3). Modules 27 and 29 were the most
significant prognosis-related lncRNA modules, which displayed the
optimal performance in survival prediction (p=0.000502 and
0.000779, respectively). Modules 27 and 29 consist of 17 and 16
lncRNAs, respectively, and the lncRNAs in them were highly
connected within the modules, indicating that they interact closely
with each other (Fig. 2). To
further investigate the mechanisms of these prognosis-related
lncRNA modules, functional pathway enrichment analysis was
performed using co-expressed genes of lncRNAs in each prognostic
lncRNA module and a prognostic lncRNA module-pathway network was
constructed (Fig. 4). We noted
that the acute myeloid leukemia pathway and pathway in cancer were
identified. Besides this, the chemokine signaling pathway (22), leukocyte transendothelial
migration pathway (23), WNT
signaling pathway (24), notch
signaling pathway (25), and T
cell receptor signaling pathway (26–28) were found to be important in AML.
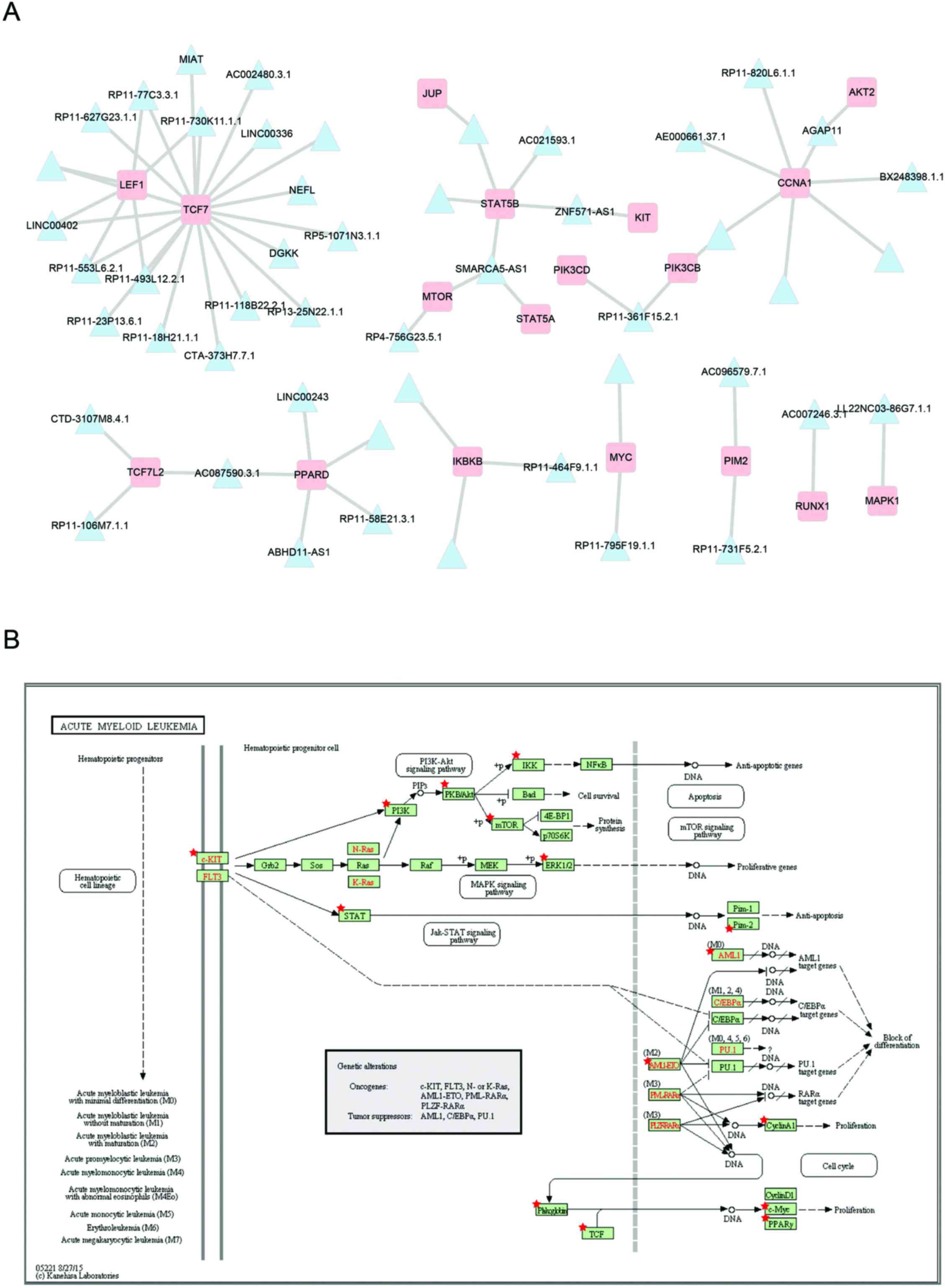
Subsequently, to examine the detailed regulatory mechanisms of the
prognostic modules, the co-expressed mRNAs of module 15 were mapped
into the AML pathway (Fig. 5). As
shown in Fig. 5A, there are 49
lncRNAs in prognosis modules co-expressed with 18 mRNAs involved in
the acute myeloid leukemia pathway (Fig. 5B). The most connected genes
lymphoid enhancer-binding factor 1 (LEF1) and transcription
factor 7 (TCF7), both map on the TCF element in the acute
myeloid leukemia pathway. Some studies have indicated that
LEF1 contributes to the pathophysiology of AML and may serve
as a novel predictor for better treatment responses (29,30). RP11-730K11.1.1 co-expressed with
both LEF1 and TCF7, suggesting that it may play an
important role in AML by regulating LEF1 and TCF7.
ZNF571-AS1 correlated with both STAT5A and KIT. KIT
is a crucial membrane protein, mutations of the KIT receptor
tyrosine kinase are involved in the constitutive activation and
development of AML and have a prognostic or possible therapeutic
impact in AML (31). The
constitutive activation of phospho-STAT5 was associated with a poor
outcome in AML, this may be via the JAK/STAT signaling pathway
(32). Thus, ZNF571-AS1 may be
involved in AML via the JAK/STAT signaling pathway by regulating
KIT and STAT5.
Although the results of the present study require
further experimental verification, they provide possible prognostic
markers for predicting patient outcome and provide further insight
into the roles of lncRNAs in AML.
Acknowledgments
This study was supported by the Health and Family
Planning Commission of Heilongjiang Province of China (no.
2014-322).
References
|
1
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: European LeukemiaNet: Diagnosis and management of acute
myeloid leukemia in adults: Recommendations from an international
expert panel, on behalf of the European LeukemiaNet. Blood.
115:453–474. 2010. View Article : Google Scholar
|
|
2
|
Schlenk RF, Döhner K, Krauter J, Fröhling
S, Corbacioglu A, Bullinger L, Habdank M, Späth D, Morgan M, Benner
A, et al: German-Austrian Acute Myeloid Leukemia Study Group:
Mutations and treatment outcome in cytogenetically normal acute
myeloid leukemia. N Engl J Med. 358:1909–1918. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marcucci G, Maharry KS, Metzeler KH,
Volinia S, Wu YZ, Mrózek K, Nicolet D, Kohlschmidt J, Whitman SP,
Mendler JH, et al: Clinical role of microRNAs in cytogenetically
normal acute myeloid leukemia: miR-155 upregulation independently
identifies high-risk patients. J Clin Oncol. 31:2086–2093. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Díaz-Beyá M, Navarro A, Ferrer G, Díaz T,
Gel B, Camós M, Pratcorona M, Torrebadell M, Rozman M, Colomer D,
et al: Acute myeloid leukemia with translocation (8;16)(p11;p13)
and MYST3-CREBBP rearrangement harbors a distinctive microRNA
signature targeting RET proto-oncogene. Leukemia. 27:595–603. 2013.
View Article : Google Scholar
|
|
5
|
Díaz-Beyá M, Brunet S, Nomdedéu J, Tejero
R, Díaz T, Pratcorona M, Tormo M, Ribera JM, Escoda L, Duarte R, et
al: Cooperative AML group CETLAM (Grupo Cooperativo Para el Estudio
y Tratamiento de las Leucemias Agudas y Mielodisplasias): MicroRNA
expression at diagnosis adds relevant prognostic information to
molecular categorization in patients with intermediate-risk
cytogenetic acute myeloid leukemia. Leukemia. 28:804–812. 2014.
View Article : Google Scholar
|
|
6
|
Schwind S, Maharry K, Radmacher MD, Mrózek
K, Holland KB, Margeson D, Whitman SP, Hickey C, Becker H, Metzeler
KH, et al: Prognostic significance of expression of a single
microRNA, miR-181a, in cytogenetically normal acute myeloid
leukemia: A Cancer and Leukemia Group B study. J Clin Oncol.
28:5257–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kapranov P, Cheng J, Dike S, Nix DA,
Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J,
Hofacker IL, et al: RNA maps reveal new RNA classes and a possible
function for pervasive transcription. Science. 316:1484–1488. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Derrien T, Johnson R, Bussotti G, Tanzer
A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG,
et al: The GENCODE v7 catalog of human long noncoding RNAs:
Analysis of their gene structure, evolution, and expression. Genome
Res. 22:1775–1789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hirano T, Yoshikawa R, Harada H, Harada Y,
Ishida A and Yamazaki T: Long noncoding RNA, CCDC26, controls
myeloid leukemia cell growth through regulation of KIT expression.
Mol Cancer. 14:902015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu J, Li CX, Li YS, Lv JY, Ma Y, Shao TT,
Xu LD, Wang YY, Du L, Zhang YP, et al: MiRNA-miRNA synergistic
network: Construction via co-regulating functional modules and
disease miRNA topological features. Nucleic Acids Res. 39:825–836.
2011. View Article : Google Scholar
|
|
11
|
Yoshimura K, Okanoue T, Ebise H, Iwasaki
T, Mizuno M, Shima T, Ichihara J and Yamazaki K: Identification of
novel noninvasive markers for diagnosing nonalcoholic
steatohepatitis and related fibrosis by data mining. Hepatology.
63:462–473. 2016. View Article : Google Scholar
|
|
12
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ivliev AE, 't Hoen PA and Sergeeva MG:
Coexpression network analysis identifies transcriptional modules
related to proastrocytic differentiation and sprouty signaling in
glioma. Cancer Res. 70:10060–10070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clarke C, Madden SF, Doolan P, Aherne ST,
Joyce H, O'Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown
J, et al: Correlating transcriptional networks to breast cancer
survival: A large-scale coexpression analysis. Carcinogenesis.
34:2300–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Tang H, Sun Z, Bungum AO, Edell ES,
Lingle WL, Stoddard SM, Zhang M, Jen J, Yang P and Wang L:
Network-based approach identified cell cycle genes as predictor of
overall survival in lung adenocarcinoma patients. Lung Cancer.
80:91–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Udyavar AR, Hoeksema MD, Clark JE, Zou Y,
Tang Z, Li Z, Li M, Chen H, Statnikov A, Shyr Y, et al:
Co-expression network analysis identifies Spleen Tyrosine Kinase
(SYK) as a candidate oncogenic driver in a subset of small-cell
lung cancer. BMC Syst Biol. 7(Suppl 5): S12013. View Article : Google Scholar
|
|
17
|
Chou WC, Cheng AL, Brotto M and Chuang CY:
Visual gene-network analysis reveals the cancer gene co-expression
in human endometrial cancer. BMC Genomics. 15:3002014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu R, Cheng Y, Yu J, Lv QL and Zhou HH:
Identification and validation of gene module associated with lung
cancer through coexpression network analysis. Gene. 563:56–62.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao Q, Liu C, Yuan X, Kang S, Miao R,
Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction
of long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu J, Yu J, Cordero KE, Johnson MD, Ghosh
D, Rae JM, Chinnaiyan AM and Lippman ME: A transcriptional
fingerprint of estrogen in human breast cancer predicts patient
survival. Neoplasia. 10:79–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
22
|
Kremer KN, Peterson KL, Schneider PA, Meng
XW, Dai H, Hess AD, Smith BD, Rodriguez-Ramirez C, Karp JE,
Kaufmann SH and Hedin KE: CXCR4 chemokine receptor signaling
induces apoptosis in acute myeloid leukemia cells via regulation of
the Bcl-2 family members Bcl-XL, Noxa, and Bak. J Biol Chem.
288:22899–22914. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gallay N, Anani L, Lopez A, Colombat P,
Binet C, Domenech J, Weksler BB, Malavasi F and Herault O: The role
of platelet/endothelial cell adhesion molecule 1 (CD31) and CD38
antigens in marrow microenvironmental retention of acute
myelogenous leukemia cells. Cancer Res. 67:8624–8632. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Staal FJT, Famili F, Garcia Perez L and
Pike-Overzet K: Aberrant Wnt signaling in leukemia. Cancers
(Basel). 8:782016. View Article : Google Scholar
|
|
25
|
Heidel FH, Arreba-Tutusaus P, Armstrong SA
and Fischer T: Evolutionarily conserved signaling pathways: acting
in the shadows of acute myelogenous leukemia's genetic diversity.
Clin Cancer Res. 21:240–248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y: Alterations in the expression
pattern of TCR zeta chain in T cells from patients with
hematological diseases. Hematology. 13:267–275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Le Dieu R, Taussig DC, Ramsay AG, Mitter
R, Miraki-Moud F, Fatah R, Lee AM, Lister TA and Gribben JG:
Peripheral blood T cells in acute myeloid leukemia (AML) patients
at diagnosis have abnormal phenotype and genotype and form
defective immune synapses with AML blasts. Blood. 114:3909–3916.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi L, Chen S, Lu Y, Wang X, Xu L, Zhang
F, Yang L, Wu X, Li B and Li Y: Changes in the MALT1-A20-NF-κB
expression pattern may be related to T cell dysfunction in AML.
Cancer Cell Int. 13:372013. View Article : Google Scholar
|
|
29
|
Fu Y, Zhu H, Wu W, Xu J, Chen T, Xu B,
Qian S, Li J and Liu P: Clinical significance of lymphoid
enhancer-binding factor 1 expression in acute myeloid leukemia.
Leuk Lymphoma. 55:371–377. 2014. View Article : Google Scholar
|
|
30
|
Metzeler KH, Heilmeier B, Edmaier KE,
Rawat VP, Dufour A, Döhner K, Feuring-Buske M, Braess J,
Spiekermann K, Büchner T, et al: High expression of lymphoid
enhancer-binding factor-1 (LEF1) is a novel favorable prognostic
factor in cytogenetically normal acute myeloid leukemia. Blood.
120:2118–2126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yohe S: Molecular genetic markers in acute
myeloid leukemia. J Clin Med. 4:460–478. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garg S, Shanmukhaiah C, Marathe S, Mishra
P, Babu Rao V, Ghosh K and Madkaikar M: Differential antigen
expression and aberrant signaling via I3/AKT, MAP/ERK, JAK/STAT,
and Wnt/beta catenin pathways in Lin-/CD38-/CD34 cells in acute
myeloid leukemia. Eur J Haematol. 96:309–317. 2016. View Article : Google Scholar
|