Introduction
Primary effusion lymphoma (PEL, also termed
body-cavity-based lymphoma) is a malignant B-cell lymphoma caused
by Kaposi's sarcoma-associated herpesvirus (KSHV, also named HHV-8)
in immunosuppressed individuals, such as AIDS patients or those
that have undergone organ transplantation (1,2). PEL
is a subtype of non-Hodgkin's lymphoma and is characterized by
lymphomatous effusions of pleural and abdominal cavities. KSHV is a
rhadinovirus of the γ-herpesvirus subfamily and is closely related
to herpesvirus Saimiri and Epstein-Barr virus (EBV). KSHV is the
causative agent of Kaposi's sarcoma and AIDS-related
lymphoproliferative disorders, such as PEL and multicentric
Castleman's disease (3). Similar
to other herpesviruses, KSHV has two life cycles (latency and lytic
replication). The KSHV genome circularizes and forms a
double-stranded DNA, the episome, in the nucleus of PEL cells
during latent infection. To establish a latent infection, KSHV
expresses several viral genes, including latency-associated nuclear
antigen (LANA), v-FLIP, v-cyclin, kaposin and microRNAs, in PEL
cells. LANA is required for the replication and maintenance of
viral DNA, and contributes to KSHV-associated oncogenesis through
interaction with cellular molecules, such as p53, Rb and GSK-3.
These viral proteins and RNA manipulate cellular signaling
pathways, including nuclear factor-κB (NF-κB), Akt, Wnt and Erk, to
maintain the malignant phenotype and ensure PEL cell survival
(4–6). Especially, NF-κB signaling is
constitutively activated in KSHV-infected PEL cells to facilitate
anti-apoptosis and growth (7–11).
KSHV alternates between lytic replication and latency by RTA
expression. RTA, encoded by the immediate-early gene ORF50, is a
critical switch molecule for initiating lytic replication.
In canonical NF-κB signaling, the NF-κB
transcription factor, which is a heterodimer of p50 and p65/RelA,
is retained in the cytosol by interaction with IκBα (12). IκBα is further regulated by the IκB
kinase (IKK) complex, consisting of IKKα, IKKβ and IKKγ/NEMO. A
stimulus, such as LPS or IL-1β, induces activation of TRAF6 through
MyD88 and IRAK, and TRAF6 induces the activation of TAK1-TAB2
complex, which phosphorylates the catalytic subunit IKKβ and
activates the IKK complex (13).
TRAF6, an E3 ubiquitin ligase, can modify IKKγ and TRAF6 itself via
K63-linked polyubiquitination, and K63-linked polyubiquitination of
IKKγ and TRAF6 contributes to the activation of IKK complex
(14). The activated IKK complex
phosphorylates Ser32 and Ser36 of IκBα. Phosphorylated IκBα is
polyubiquitinated by ubiquitin ligase E3 and subsequently
destabilized by proteasomal degradation. This degradation of IκBα
leads to nuclear translocation of NF-κB and the NF-κB-dependent
transcriptional activation.
Garlic (Allium sativum L.) is widely used in
traditional herbal remedies and alternative medicine. Garlic oil
from fresh garlic contains allyl sulfides, including diallyl
sulfide (DAS), diallyl disulfide (DAD), diallyl trisulfide (DAT),
and other allyl polysulfides (15). The proportions of allyl sulfides in
garlic oil are ~6% DAS, 30% DAD, 40% DAT and 24% other analogs
(16). Allyl sulfides have many
biological functions, including suppression of inflammation,
upregulation of detoxification, enhancement of histone acetylation,
generation (or reduction) of reactive oxygen species (ROS), and
induction of endoplasmic reticulum (ER) stress (17,18).
There have been many reports of anticancer effects of these
compounds against a variety of cancers, including prostate, lung,
breast, and colon cancer cells (19–22).
In addition, DAT directly produces ROS through the homolytic
cleavage of intramolecular trisulfide bonds, and DAT-mediated ROS
induces oxidative and ER stress, causing apoptosis of various
cancer cell types (20–23). Recently, DAT has been shown to
affect cellular signaling pathways including Akt, NF-κB, Jnk and
Erk, in tumor cells (24,25).
PEL is an aggressive lymphoma caused by KSHV, and is
resistant to chemotherapy regimens, such as CHOP and R-CHOP
(26). Therefore, the development
of novel and effective drugs for PEL is required. Regarding
biological properties of allyl sulfides, these could be novel
compounds used with molecular target drugs for the treatment of
PEL. The anti-cancer effect of allyl sulfides against PEL remains
unknown; we therefore investigated whether allyl sulfides kill PEL
cells and the underlying molecular mechanism thereof.
Materials and methods
Cells and reagents
KSHV-positive PEL cell lines (BC2, BC3, BCBL1 and
HBL6) and KSHV-negative lymphoma cell lines (Ramos, BJAB and DG75)
were maintained in RPMI-1640 medium supplemented with 10% fetal
calf serum. DAS (Wako, Osaka, Japan), DAD (Tokyo Chemical Industry,
Tokyo, Japan), DAT (LKT Laboratories, St. Paul, MN, USA) were
dissolved in dimethyl sulfoxide (DMSO).
Cell viability assay
Cells were seeded on 96-well plates at
2.5×104 cells/well in 100 μl of medium with or without
various concentrations of DAS, DAD or DAT and then incubated at
37°C for 24 h. The number of viable cells was estimated using a
Cell Counting kit-8 (Dojindo, Tokyo, Japan) as described previously
(27). The optical density at 450
nm of each sample was measured using a microplate spectrophotometer
(Tecan M200; Tecan, Kanagawa, Japan) and expressed as a percentage
of the value in untreated cells (defined as 100%). Data are shown
as the means ± SEM of three independent experiments.
Western blotting and antibodies
Western blotting was performed as described
previously (28). Primary
antibodies used in these experiments included those against
Ser32/36-phospho-IκBα, Ser176/180-phospho-IKKα/β, caspase-3, -7,
-8, -9 and PARP (Cell Signaling Technology, Danvers, MA, USA),
IκBα, p65, and p21Cip1 (BD Biosciences, Franklin Lakes,
NJ, USA), β-actin, histone H2B, K-bZIP and S-tag (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). FK2 that we established
previously (29) was used to
detect polyubiquitin molecules.
Caspase assay
To measure caspase activity, 1.0×105 BC3
PEL cells/well were added to 1 ml of medium and incubated with DAT
for 3 h. Activities of caspase-8 and -9 in cell lysates were
measured using a caspase-Glo assay kit (Promega, Madison, WI, USA)
with luciferin-conjugated LETD or LEHD polypeptide, respectively,
according to the manufacturer's instructions. Luminescence was
detected using a GloMax 20/20 luminometer (Promega). The caspase
activity in untreated cells was defined as 1.0 relative light
unit.
Immunofluorescence (IF) analysis
Prior to IF analysis, BCBL1 cells were treated with
10 μM DAT for 18 h and fixed in methanol on glass slides followed
by incubation with primary antibodies for 1 h. After washing, the
cells were incubated with Alexa Fluor 594-conjugated donkey
anti-mouse IgG or Alexa Fluor 488-conjugated donkey anti-rabbit IgG
(Invitrogen, Carlsbad, CA, USA). To stain the nuclei, cells were
incubated in 4 ng/ml of DAPI in PBS during binding of the secondary
antibody. Immunofluorescence images were obtained by fluorescence
microscopy (IX71; Olympus, Tokyo, Japan).
Nuclear protein extraction
Harvested cells were incubated with hypotonic buffer
containing 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM
EGTA, 0.1 mM PMSF, and 1 mM DTT on ice for 15 min, and then cells
were lysed by addition of NP-40 (final concentration, 0.6%) and
heavy agitation with a vortex mixer. Cell lysates were centrifuged
at 15,000 rpm for 30 sec at 4°C. The nuclear pellets were rinsed
with the hypotonic buffer supplemented with 0.6% NP-40. The
obtained nuclear extracts were lysed in SDS-PAGE sample buffer.
Luciferase reporter assay
BC3 cells (1×105) were transfected with 2
μg of NF-κB-luciferase reporter (pGL4.32) and 1 μg of pSV-β-Gal
(both from Promega) used as an internal control with 9 μg
polyethyleneimine. Transfected cells were incubated in medium with
various concentrations of DAT for 6 h. Cells were resuspended in
0.1 ml of passive lysis buffer (Promega) for luciferase assays.
Luciferase activity was measured with a GloMax 20/20 luminometer.
The luciferase activity of DAT-untreated cells was defined as 1.0
relative light unit.
Reverse transcription-polymerase chain
reaction (RT-PCR) and real-time RT-PCR
Total RNA was purified and extracted from
1×106 cells using an Illustra RNAspin Mini RNA Isolation
kit (GE Healthcare, Buckinghamshire, UK). First-strand cDNA was
synthesized from 20 ng of total RNA using a ReverTra Ace qPCR RT
kit (Toyobo, Osaka, Japan). To quantify cDNA, PCR was performed
using GoTaq Flexi DNA polymerase (Promega). The PCR products were
analyzed by electrophoresis on 2% agarose gels and staining with
ethidium bromide. The nucleotide sequences of oligonucleotides used
for RT-PCR primers were as follows: IKKα forward, 5′-AGA CGT CAG
GGA GAC TTG ATG-3′ and reverse, 5′-ACT GGA TCC TAC AAG AGA GCG-3′;
IKKβ forward, 5′-AGG TGC CAT CCT CAC CTT GC-3′ and reverse, 5′-AAT
GTC CAC CTC ACT CTT CTG CC-3′; IKKγ forward, 5′-AGT TGC AGG TGG CCT
ATC ACC-3′ and reverse, 5′-CTC ATG TCC TCG ATC CTG GC-3′; TRAF6
forward, 5′-CAG GGG TAT AGC TTG CCC TCA C-3′ and reverse, 5′-TGG
AAC GTG TGG ATT CCC AG-3′; GAPDH forward, 5′-TGA CCA CAG TCC ATG
CCA TC-3′ and reverse, 5′-GGG GAG ATT CAG TGT GGT GG-3′.
For quantification of cDNA, Real-time RT-PCR was
performed as described previously (10). Briefly, total RNA was purified and
extracted from 1×106 cells and first-strand cDNA was
synthesized from 20 ng of total RNA. To quantify cDNA, SYBR-Green
real-time PCR was performed using the MiniOpticon real-time PCR
(Bio-Rad, Hercules, CA, USA). The sequences of oligonucleotides
used for primers were as follows: IκBα forward, 5′-AGC TTT TGG TGT
CCT TGG GTG-3′ and reverse, 5′-CTG TTG ACA TCA GCC CCA CAC-3′; RTA
forward, 5′-ATA ATC CGA ATG CAC ACA TCT TCC ACC AC-3′ and reverse,
5′-TTC GTC GGC CTC TCG GAC GAA CTG A-3′; K8.1 forward, 5′-TCC ACA
CAG ATT CGC ACA GA-3′ and reverse, 5′-AAT GCG AAC GAT ACG TGG
GA-3′. The primer set for GAPDH forward, 5′-GAG TCA ACG GAT TTG GTC
GT-3′ and reverse, 5′-GAC AAG CTT CCC GTT CTC AG-3′ was used as an
internal control for normalization. For quantification, the
expression level of each gene was normalized to that of the GAPDH
gene.
Transfection and plasmids
293/TLR4-MD2-CD14 (293/TLR4) cells were purchased
from InvivoGen (San Diego, CA, USA). The 293 or HeLa cells seeded
at 2×106 cells/10 cm dish were transfected using the
Chen-Okayama calcium phosphate procedure (30) with 10 μg of plasmid, and harvested
48 h after transfection.
The open reading frames (ORFs) of human ubiquitin,
IKKα, IKKβ, IKKγ and TRAF6 were amplified by PCR. All constructs
were verified by DNA sequencing. For eukaryotic expression, the
respective PCR fragments were cloned into the pCI-neo-Flag,
pCI-neo-T7, pCI-neo-Myc or pCI-neo-S-tag vector, which had been
generated by inserting oligonucleotides encoding three repeats of a
Flag-tag sequence, three repeats of a T7-tag sequence, a Myc-tag
sequence, and two repeats of an S-tag (S-tag peptide),
respectively, into the pCI-neo mammalian expression vector
(Promega).
Immunoprecipitation
Immunoprecipitation assays to detect
polyubiquitination of IκBα were performed according to the methods
described by Saji et al (10). BC3 cells (1×107) were
lysed in 1 ml RIPA buffer. Cell lysates were incubated with 5 μg of
anti-IκBα antibody. Immunoprecipitated IκBα was immunoblotted with
FK2 (anti-polyubiquitin) or anti-IκBα antibody. Immunoprecipitation
assays to detect the interactions of IKKα, IKKβ and IKKγ were
performed, as described previously (5).
Measurement of proteasome activity
BC3 cells (1×106) were lysed in 0.2 ml of
buffer containing 50 mM Tris-HCl (pH 7.6), 1 mM MgCl2,
0.1 mM EDTA, 1% glycerol, 1 mM DTT, 0.2 mM ATP, 0.2% NP-40, and
homogenized with 27 G needles and 1 ml disposable syringes. The
chymotrypsin-like activity of the proteasome in cell lysate was
assessed with the fluorogenic peptide,
Suc-Leu-Leu-Val-Tyr-4-methylcoumaryl-7-amide (MCA) (Peptide
Institute, Osaka, Japan). The cell lysates (5 μl) were incubated in
95 μl of reaction buffer containing 50 mM Tris-HCl (pH 7.8), 10 mM
MgCl2, 1 mM DTT, 2 mM ATP, 0.1 mM
Suc-Leu-Leu-Val-Tyr-MCA. The fluorescence intensity owing to AMC
(excitation, 380 nm; emission, 460 nm) was determined using a
microplate spectrofluorometer (Infinite M200; Tecan, Kawasaki,
Japan).
Real-time PCR measurement of viral
load
Real-time PCR was performed by the method described
previously (10). BCBL1 cells
(2×105) were treated with 3 mM n-butyrate and DAT for 48
h, and the culture media were harvested. To obtain only enveloped
and encapsulated viral genomes, media were incubated with DNase I,
and viral DNA was purified using a QIAamp DNA blood mini kit
(Qiagen, Hilden, Germany). To quantify viral DNA, SYBR Green
real-time PCR was performed by MiniOpticon real-time PCR (Bio-Rad).
To generate a standard curve for cycle threshold versus genomic
copy number, the pCIneo-KSHV ORF50/RTA plasmid was serially diluted
to known concentrations in the range of
101–106 plasmid molecules/μl. Each PCR
mixture contained 1 μl viral or standard DNA, 0.5 μM ORF50 primers,
10 μl SYBR Master Mix Plus (Bio-Rad), and H2O in a total
volume of 20 μl. The following primers were used to amplify a
140-bp amplicon internal to the ORF50 sequence: ORF50/RTA forward,
5′-ATA ATC CGA ATG CAC ACA TCT TCC ACC AC-3′ and reverse, 5′-TTC
GTC GGC CTC TCG GAC GAA CTG A-3′. The PCR conditions were as
follows: 95°C for 2 min, followed by 95°C for 5 sec and 62°C for 20
sec, repeated for 40 cycles.
Xenograft mouse model of PEL
CB17 SCID female mice aged 5 weeks were purchased
from CLEA Japan (Tokyo, Japan). Mice were allowed to feed ad
libitum on sterilized laboratory chow and water. Mice were
injected intraperitoneally with 5×107 BCBL1 cells before
1 week of DAT administration. The DAT dissolved in corn oil or corn
oil alone was administered into the intraperitoneal region at a
dose of 5 mg/kg body weight each day for 3 weeks. Mice were
observed and body weight was measured each day for 3 weeks. All
mice were sacrificed on day 21, and the ascites and organs were
collected. The ascites collected from each mouse was centrifuged to
determine the tumor weight. Genomic DNA of tumor cells in ascites
was purified using SepaGene (EIDIA, Tokyo, Japan) and subjected to
real-time PCR to quantify viral DNA. All experiments were carried
out in accordance with the Code of Ethics of the World Medical
Association (Declaration of Helsinki) and the guiding principles
for the care and use of laboratory animals in Kyoto Pharmaceutical
University (KPU). Animal studies were approved by the Experimental
Animal Research Committee at KPU.
Results
Cytotoxic effects of DAT on PEL
cells
First, we examined the cytotoxic effects of allyl
sulfides (DAS, DAD and DAT) on KSHV-infected PEL cell lines (BC3,
BCBL1, HBL6 and BC2) and KSHV-uninfected lymphoma cell lines (Ramos
and DG75). These B-lymphoma cells were cultured in the presence of
DAS, DAD or DAT for 24 h, and the cytotoxicity was evaluated by
analyzing the viability of allyl sulfide-treated versus untreated
cells (Fig. 1). DAS and DAD
treatments slightly decreased the viability of PEL cells compared
to KSHV-uninfected cells (Fig. 1A and
B) whereas DAT treatment selectively prevented the
proliferation of PEL cell lines at lower concentrations than
required for KSHV-uninfected Ramos and DG75 cell lines (Fig. 1C). In particular, treatment with 20
μM DAT showed marked antiproliferative effects on PEL cell lines
with no influence on KSHV-uninfected cell lines. The cytotoxic
effects of DAT on B-lymphoma cells are summarized in Table I. As DAT showed strong
antiproliferative activities against PEL, we focused on the
cytotoxic effects of DAT and analyzed the underlying molecular
machinery.
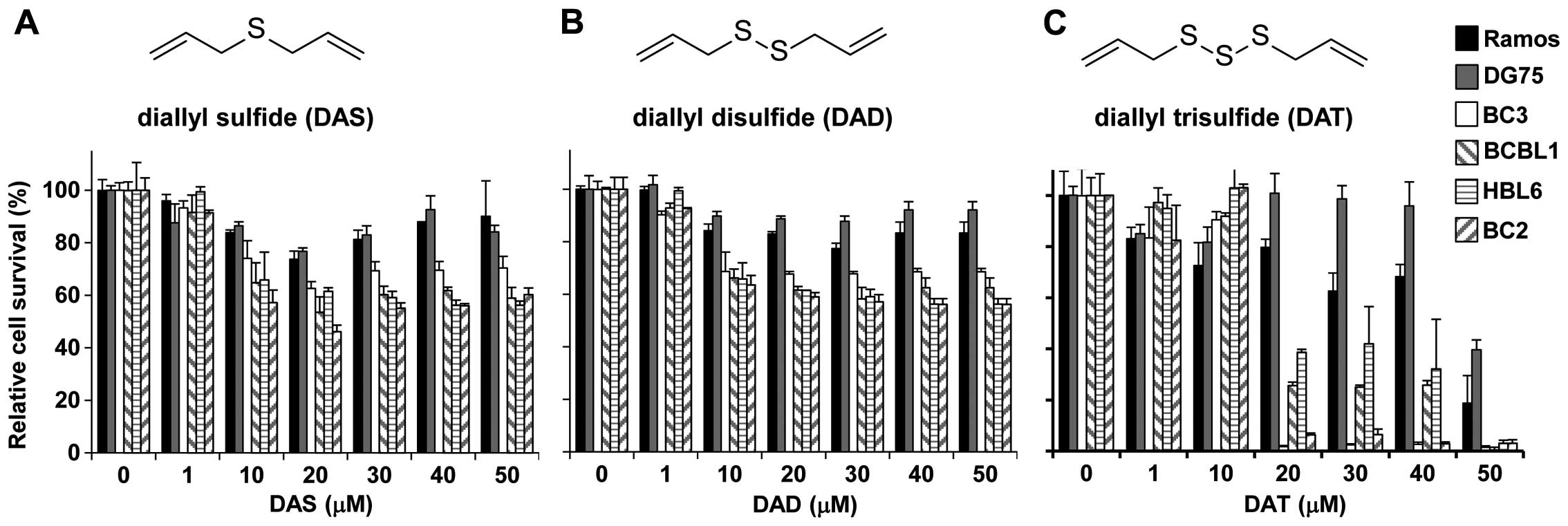 | Figure 1Cytotoxic effects of diallyl sulfide
(DAS), diallyl disulfide (DAD), and diallyl trisulfide (DAT) on PEL
cells and KSHV-uninfected lymphoma cells. (A–C) Show the structures
and cytotoxic effects of DAS, DAD and DAT, respectively.
KSHV-positive PEL cells (BC3, BCBL1, BC2 and HBL6 cells) and
KSHV-uninfected lymphoma cells (Ramos and DG75 cells) were
incubated with various concentrations of DAS, DAD, and DAT for 24 h
and subjected to cell viability assays. For each cell type,
viability was assessed in three replicate wells. The optical
density at 450 nm was measured, and the values of the respective
untreated cells were defined as 100%. Standard deviations were
determined by analysis of data from three independent experiments,
and are indicated by error bars. |
 | Table ICytotoxic effects of DAT on
B-lymphoma cells. |
Table I
Cytotoxic effects of DAT on
B-lymphoma cells.
| BC3 | BCBL1 | HBL6 | BC2 | Ramos | DG75 |
|---|
| CC50
(μM) | 13.7±0.8 | 15.5±1.0 | 17.7±0.6 | 14.6±0.4 | 43.4±1.4 | 48.0±0.9 |
DAT induces apoptosis through the
activation of caspases in PEL cells
We next investigated whether the cytotoxic effects
of DAT were due to apoptotic cell death. Apoptosis is induced by
the activation of executioner caspases, including caspase-3 and -7,
which have been previously activated via either an intrinsic
pathway (caspase-9) or an extrinsic pathway (caspase-8). We
monitored the cleavage (i.e., activation) of caspase-3, -7, -8 and
-9 by immunoblotting of lysates prepared from PEL and Ramos cells
cultured with DAT (Fig. 2A).
Active caspase-3, -7 and -9 were detected in BC3 and BCBL1 cells
that had been treated with DAT for 6 and 12 h. Further, the amounts
of cleaved PARP were increased in DAT-treated BC3 and BCBL1 cells.
However, on immunoblotting analysis, active caspase-8 was slightly
detected in BC3 and BCBL1 cells. In contrast, neither the cleavage
of caspase nor the accumulation of cleaved PARP was detected in
DAT-treated Ramos cells. In addition to the activation of caspase,
p21Cip1 was upregulated in DAT-treated cells. To obtain
further evidence of the activation of caspase-9, we measured the
peptidase activities of caspase-8 and -9 in BC3 cells pretreated
with DAT (Fig. 2B). Compared to
treatment with vehicle control, treatment with DAT increased
caspase-8 and -9 activities by 1.2- and 1.6-fold, respectively.
There was no significant difference in caspase-8 between
DAT-treatment and control. These data indicate that DAT inhibited
the growth of PEL cells by cell cycle arrest, leading to
caspase-9-dependent apoptosis. We adopted different treatment times
and drug concentrations as shown in Fig. 2. The reasons of treatment times are
as follows. Activations of executioner caspases (caspase-3 and -7)
and PARP cleavage occur later in the apoptosis cascade, while
activations of initiator caspases (caspase-9 and -8) occur early in
the apoptosis. In fact, strong activation of caspase-9 and PARP
cleavage were detected in cells treated with DAT for 6 and 12 h,
respectively (Fig. 2A). Regarding
drug concentrations, more DAT were necessary for western blotting
to detected cleaved molecules (i.e., activated forms) because
western blot analysis has low detection sensitivity as compared
with peptidase assay.
DAT suppresses NF-κB signaling through
the stabilization of IκBα
Several signaling pathways are activated in PEL
cells to maintain malignant potential and enhance cell survival.
Especially, the constitutive and/or transient activation of NF-κB
signaling is necessary for PEL to achieve the establishment of KSHV
infection, survival of infected cells, and viral replication
(4,7–11).
To obtain insight into the machinery by which DAT induces apoptosis
of KSHV-infected PEL cells, we analyzed the effect of DAT treatment
on NF-κB signaling. PEL and KSHV-uninfected Ramos cells were
incubated in medium with 10 μM DAT, expression of IκBα was
elucidated by western blot analysis. DAT treatment significantly
increased the expression of IκBα in PEL cells, whereas,
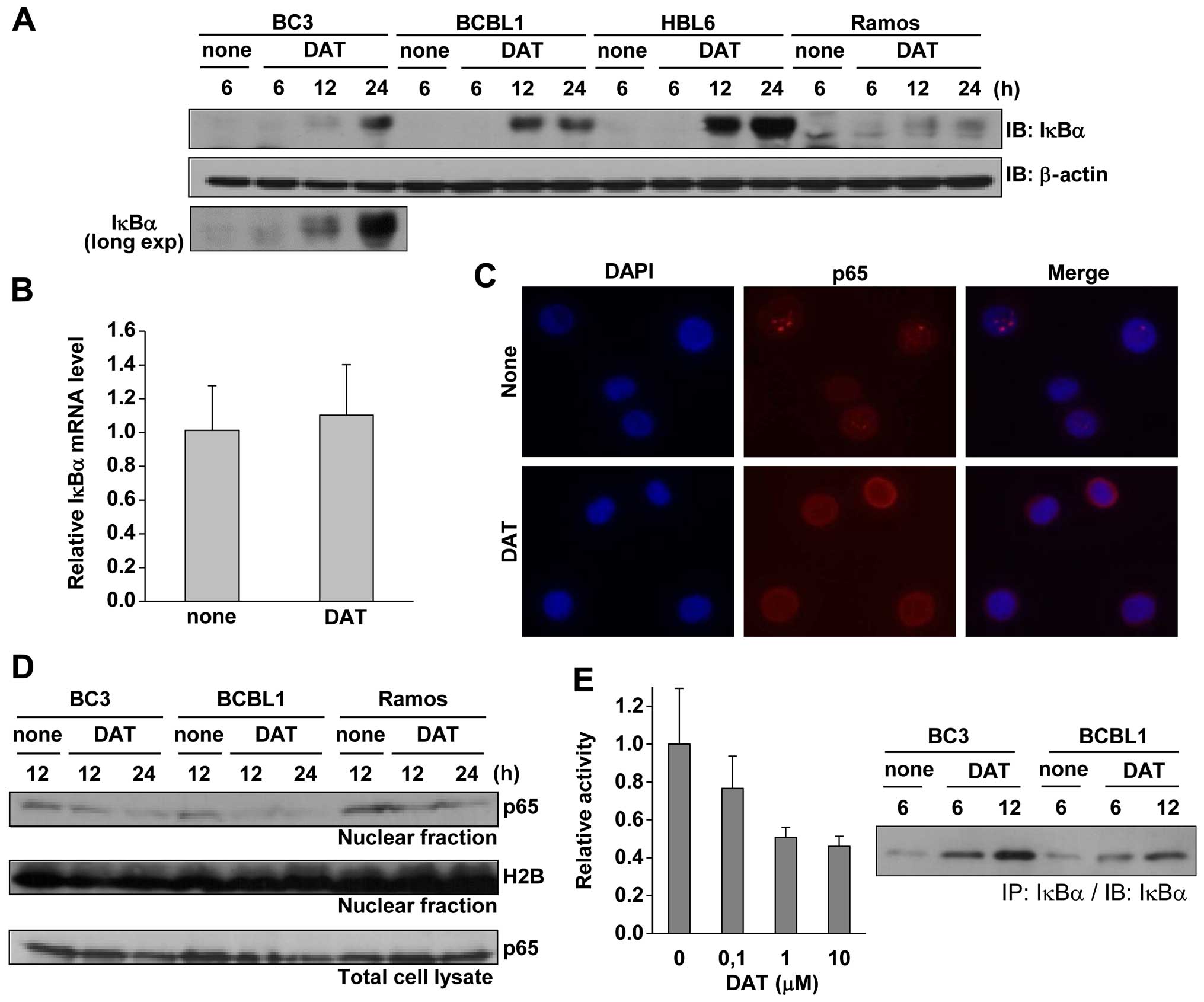
upregulation of IκBα was not detected in Ramos cells (Fig. 3A). The long exposure image is
shown, indicating the IκBα upregulation in BC3 treated with DAT for
6 and 12 h. To determine whether DAT-mediated upregulation of IκBα
is due to transcriptional activation of IκBα, we measured the
expression of IκBα mRNA in PEL cells treated with DAT by real-time
RT-PCR (Fig. 3B). The result
indicated that DAT treatment had no influence on IκBα mRNA in
BCBL1, suggesting that DAT induces the stabilization of IκBα. As
DAT showed increased expression of IκBα, we next examined the
suppression of NF-κB nuclear translocation and the downregulation
of NF-κB-dependent transcriptional activity by DAT treatment. IF
analysis showed that compared to untreated controls, DAT treatment
induced cytosolic localization of p65 in BCBL1 cells (Fig. 3C). The amount of p65 in the nucleus
decreased in BC3 and BCBL1 cells treated with DAT for 12 and 24 h
(Fig. 3D), as compared to Ramos
cells, while DAT did not alter the total amount of p65 in all cell
lines (Fig. 3D). In addition, we
performed a reporter assay using the NF-κB reporter plasmid to
confirm the suppression of NF-κB in BC3 cells by DAT-treatment. As
expected, DAT treatment suppressed NF-κB transcriptional activity
of BC3 cells in a concentration-dependent manner (Fig. 3E). Western blot analysis for
detection of immunoprecipitated IκBα using lysates from DAT-treated
BC3 and BCBL1 is also shown. Data show that DAT-treatment for 6 h
significantly induced IκBα upregulation in PEL cells. These data
indicated that DAT treatment suppressed NF-κB signaling through the
stabilization of IκBα in PEL cells.
DAT induces stabilization of IκBα by
inhibiting phosphorylation of IκBα
The mechanism of IκBα degradation consists of three
sequential steps: first, the phosphorylation of IκBα by IKK
complex; second, polyubiquitination of phosphorylated IκBα by E3
ubiquitin ligase; and third, degradation of polyubiquitinated IκBα
by 26S proteasome. We attempted to determine which step is
influenced by DAT treatment, leading to the stabilization of IκBα.
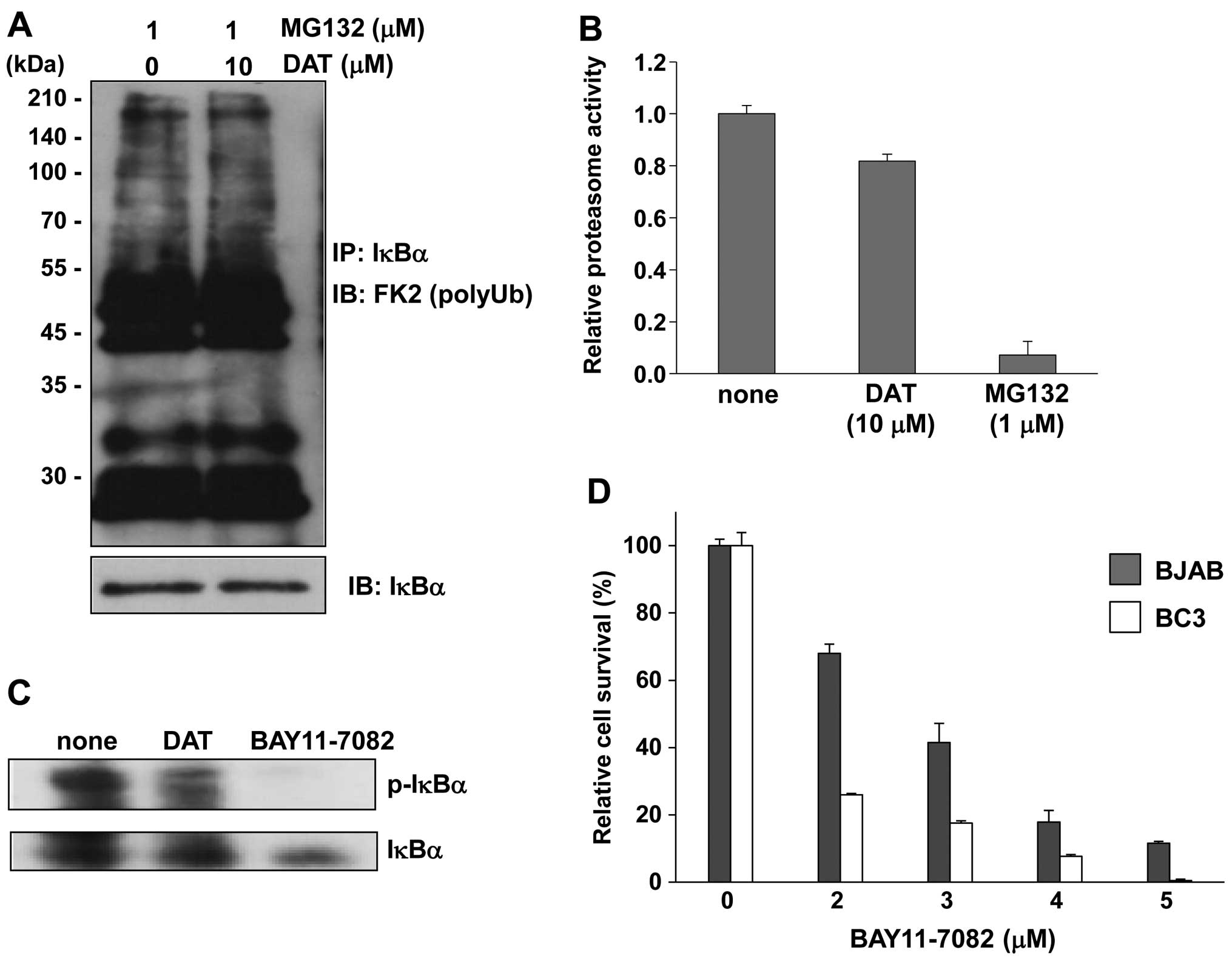
First, we monitored polyubiquitination of IκBα (Fig. 4A). To detect polyubiquitination of
IκBα, immunoprecipitated IκBα from DAT and MG132-treated BC3 cell
lysate was subsequently probed by immunoblotting with an
anti-polyubiquitin antibody (FK2). However, there was no difference
in amount of polyubiquitination of IκBα between DAT-treated and
untreated cells. Next, to examine whether DAT inhibited the
degradation of IκBα by the 26S proteasome, we measured the
chymotrypsin-like activity of the proteasome. In BC3 cells treated
with DAT for 24 h, the proteasome activity decreased by ~20%
compared to that in untreated cells (Fig. 4B). The proteasome inhibitor MG132
was used as a positive control. We also investigated the effects of
DAT on IκBα phosphorylation in PEL cells. When BC3 cells were
treated with DAT, the amount of phosphorylated IκBα (p-IκBα)
protein was significantly decreased compared to untreated cells
(Fig. 4C). BAY11-7082, which
inhibits IKK and IκB phosphorylation, was used as a positive
control. We evaluated the cytotoxic effect of BAY11-7082 on PEL to
confirm the contribution of IκBα phosphorylation to apoptotic
activity on PEL. Treatment with BAY11-7082 markedly decreased the
viability of BC3 PEL cells compared to KSHV-uninfected BJAB cells
(Fig. 4D). Thus, our data
demonstrated that DAT stabilized IκBα through inhibition of IκBα
phosphorylation. Furthermore, DAT-mediated suppression of NF-κB
signaling through inhibition of IκBα phosphorylation can be a
trigger for apoptosis of PEL cells.
DAT suppresses the phosphorylation of
IKKβ through downregulation of TRAF6
We demonstrated that DAT stabilized IκBα through
inhibition of IκBα phosphorylation in PEL cells. To determine
whether DAT-mediated phosphorylation of IκBα is due to quantitative
or qualitative changes in IKK, we examined the mRNA expression,
complex formation and phosphorylation state of IKK in DAT-treated
PEL. The results indicated that DAT treatment did not affect the
levels of IKKα, IKKβ or IKKγ mRNA in BCBL1 or Ramos cells (Fig. 5A). We next examined the
complexation of IKK in DAT-treated cells. HeLa cells were
transfected with Flag-IKKα, T7-IKKβ and S-tag-IKKγ expression
plasmids, and treated with DAT. Cell lysates were subjected to
coprecipitation assay using S protein-immobilized beads. However,
there were no changes in complex formation of IKK associated with
DAT treatment (Fig. 5B). Next, we
examined whether DAT suppresses the phosphorylation of endogenous
IKKα or IKKβ in PEL, HeLa and 293 cells. However, we could not
detect the phosphorylation signal of endogenous IKKα/β, because the
phosphorylation and expression levels of IKKα/β in normal cultured
cells were too low. Therefore, we used 293/TLR4 cells
constitutively expressing TLR4. 293/TLR4 cells transfected with
either Flag-IKKα or T7-IKKβ cultured with or without DAT-containing
medium in the presence of LPS to activate the NF-κB signal. When
Flag-tagged IKKα-transfected cells were treated with LPS, small
amounts of Ser176 in exogenous IKKα were phosphorylated, and DAT
treatment suppressed this phosphorylation (Fig. 5C). On the contrary, in T7-tagged
IKKβ-transfected cells, large amounts of IKKβ were phosphorylated
in an LPS-independent manner, and DAT treatment strongly suppressed
the phosphorylation of Ser180 at T7-IKKβ (Fig. 5D). That is, DAT suppressed the IKK
complex by inhibiting the phosphorylation of IKKβ, which is the
catalytic subunit of the IKK complex and directly phosphorylates
IκBα for degradation. As the ubiquitin ligase, TRAF6, which is
self-activated by TRAF6 polyubiquitination, induces IKKβ
phosphorylation via activation of TAK1-TAB2 complex, we compared
the self-polyubiquitination of TRAF6 in the presence or absence of
DAT. HeLa cells transfected with T7-TRAF6 and Myc-ubiquitin were
cultured with or without DAT, and cell extracts were assayed by
coimmunoprecipitation assay. Immunoprecipitated T7-TRAF6 was
immunoblotted with anti-T7 and anti-Myc antibody to detect TRAF6
and polyubiquitinated TRAF6, respectively. DAT treatment slightly
reduced the polyubiquitination of TRAF6 (Fig. 5E, left panel), but significantly
reduced the amount of TRAF6 protein (Fig. 5E, right panel). Therefore, we
compared the destabilization of TRAF6 in the presence or absence of
DAT. S-tag-TRAF6-transfected 293/TLR4 or HeLa cells were incubated
with DAT, and TRAF6 was detected by immunoblotting with an
anti-S-tag antibody. The results indicated that DAT treatment for 3
and 6 h induced destabilization of TRAF6 in 293/TLR4 (Fig. 5F, upper panel) and HeLa cells
(Fig. 5F, lower panel). In
addition, the proteasome inhibitor, MG132, overcame the DAT-induced
destabilization of TRAF6. In Fig.
5A, we confirmed that DAT did not affect the expression of
TRAF6 mRNA in cells. These data indicate that DAT induces
degradation of TRAF6 by the proteasome, and DAT-mediated
downregulation of TRAF-6 suppresses the phosphorylation of IKKβ and
activation of IKK complex. Furthermore, DAT-mediated suppression of
IKK induces the suppression of IκBα phosphorylation for
polyubiquitination and proteasomal degradation of IκBα.
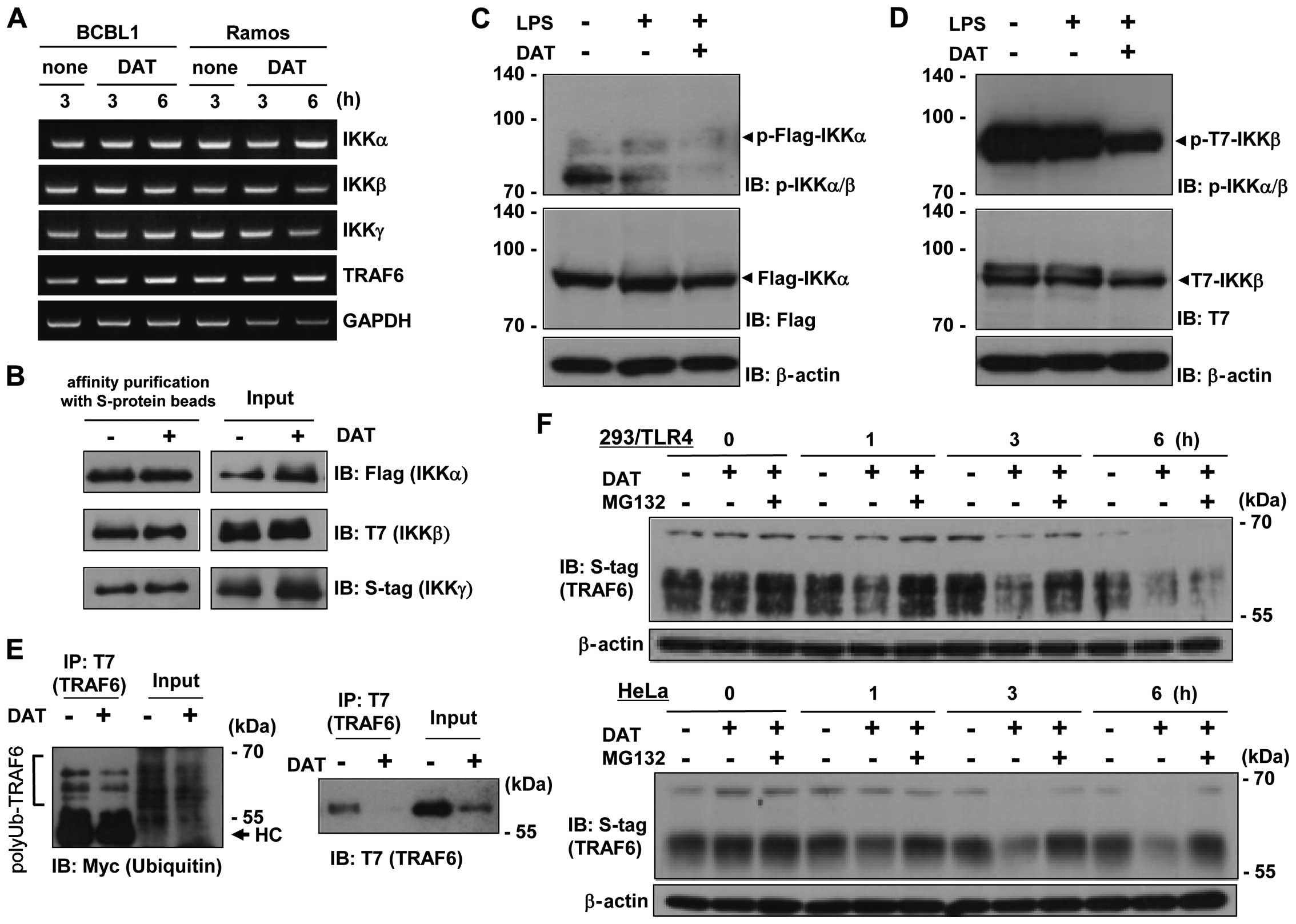 | Figure 5Inhibition of IKKα phosphorylation
through destabilization of TRAF6 by DAT. (A) RT-PCR to measure the
mRNA expression of IKKα, IKKβ, IKKγ and TRAF6 in DAT-treated PEL.
Cells were cultured with 20 μM DAT for 3 or 6 h, and the mRNA were
detected from extracted total RNA by RT-PCR. (B) Influence of DAT
on IKK complex (IKKα-IKKβ-IKKγ) formation. HeLa cells were
transfected with Flag-IKKα, T7-IKKβ and S-tag-IKKγ plasmids and
cultured for 24 h. Transfected cells were treated with or without
10 μM DAT for 24 h. The cell extracts were subjected to affinity
purification using S-protein-immobilized agarose beads, and
purified proteins (S-tag-IKKγ) were subjected to western blotting.
(C) Inhibition of IKKα phosphorylation by DAT. 293/TLR4 cells were
transfected with 6 μg of Flag-IKKα expression plasmid. Twenty-four
hours after transfection, cells were treated with or without 20 μM
DAT for 6 h in DMEM containing 40 ng/ml lipopolysaccharide (LPS).
Harvested cells were subjected to immunoblotting using
anti-Ser176/180 phospho-IKKα/β antibody to detect
Ser176-phosphorylated Flag-IKKα. The anti-Ser176/Ser180
phospho-IKKα/β antibody recognizes Ser176-phosphorylated IKKα and
Ser180-phosphorylated IKKβ. (D) Inhibition of IKKβ phosphorylation
by DAT. 293/TLR4 cells were transfected with T7-IKKβ, and cells
were treated with DAT and LPS for 6 h. Cells were subjected to
immunoblotting using anti-Ser176/180-phospho-IKKα/β to detect
Ser180-phosphorylated T7-IKKβ. (E) Effect of DAT on
polyubiquitination of TRAF6. HeLa cells were transfected with 3 μg
of T7-TRAF6 and 3 μg of Myc-ubiquitin (Ub) expression plasmids, and
cultured with 20 μM DAT in DMEM for 24 h. Cell extracts were
incubated with 10 μl of anti-T7 antibody-immobilized agarose beads
for 1 h. To detect polyubiquitination of TRAF6, immunoprecipitates
(T7-TRAF6) were subjected to SDS-PAGE followed by blotting with
anti-T7 (to detect TRAF6) and Myc (to detect polyubiquitin) mouse
antibodies. The arrowhead indicates Ig heavy chain. (F)
Proteasome-dependent destabilization of TRAF6 by DAT. 293/TLR4 or
HeLa cells were transfected with 6 μg of S-tag-TRAF6 and cultured
for 24 h. Transfected cells were cultured with 20 μM DAT and 10 μM
MG132 in DMEM containing 25 μg/ml cycloheximide for 0, 1, 3 or 6 h,
and lysed in SDS-PAGE sample buffer. To determine the amount of
TRAF6 protein, lysates of 293/TLR4 (upper panel) or HeLa cells
(lower panel) were analyzed by immunoblotting using an anti-S-tag
rabbit polyclonal antibody. |
DAT inhibits KSHV replication in PEL
NF-κB signaling is essential for replication of KSHV
(10,11). Therefore, we investigated whether
DAT suppresses KSHV lytic replication in PEL cells (Fig. 6A). The results showed that DAT
suppressed viral particle production at low concentrations, which
did not affect BCBL1 cell growth (Fig.
1). Next, to investigate which step of viral replication is
prevented by DAT we examined the effect of DAT on lytic gene
expression for viral replication. DAT treatment decreased mRNA
expression of RTA (immediate-early gene) and K8.1 (late gene) in
BCBL1 cells lytically induced by n-butyrate (Fig. 6B). DAT treatment was suggested to
influence the early stage of KSHV replication, i.e., viral gene
transcription, in PEL cells. These results indicate that NF-κB
activity is required for both KSHV-infected cell survival and viral
replication in PEL cells.
Treatment with DAT suppresses the
development of PEL in SCID mice
As DAT showed selective cytotoxicity on PEL cell
lines (Fig. 1), we next
investigated whether DAT treatment exerted cytotoxic effects
against xenograft PEL cells in SCID mice. BCBL1 cells were injected
intraperitoneally into SCID mice 1 week prior to commencement of
DAT administration. DAT or vehicle (corn oil) was injected
intraperitoneally each day for 21 days. The mice with and without
DAT administration showed significantly different gross appearance
(Fig. 7A): the abdomen of corn
oil-treated PEL-xenografted mice (control mice) showed expansion,
whereas DAT-treated mice had an apparently normal body shape.
Moreover, the body-weight increase of DAT-treated mice was less
marked than that of control mice (Fig.
7B). Necropsy revealed that the spleens of control mice showed
distention compared to those of DAT-treated mice (Fig. 7C), whereas there were no
significant morphological differences in other organs between
DAT-treated and -untreated mice. The weight of the spleen in the
DAT-treated group was ~0.07 g, which was lower than that of the
corn oil-treated group (Fig. 7D).
As the average weight of the spleen in normal 6–7-week-old SCID
mice is ~0.06 g (data not shown), the spleens of control mice were
considerably larger than those of normal and DAT-treated mice. The
weight of tumor cells in ascites in the DAT-treated group was
significantly less than that of control group (Fig. 7E). Real-time PCR indicated that
ascites and spleen tumor cells were infected with KSHV in corn
oil-treated mice, and DAT treatment markedly decreased KSHV DNA in
ascites and spleen compared to control (Fig. 7F and G). IF analysis showed that
the tumor cells in ascites from control mice expressed LANA, a
marker of latent KSHV infection (Fig.
7H). These results indicated that xenografted BCBL1 developed
in ascites of corn oil-treated control mice, and DAT prevented
BCBL1 development in ascites.
 | Figure 7In vivo effects of DAT on
development of PEL cells in SCID mice. (A) Photograph showing
DAT-treated (right) and DAT-untreated (left) SCID mice on day 21
after commencement of DAT administration. SCID mice were injected
intraperitoneally with 5×107 BCBL1 cells 1 week before
commencement of DAT administration. DAT dissolved in corn oil
(DAT-treated) or corn oil alone (DAT-untreated) was administered
intraperitoneally at a dose of 5 mg/kg body weight each day for 21
days. (B) The body weight changes of BCBL1-xenografted SCID mice
for 3 weeks every day from commencement of intraperitoneal DAT or
corn oil administration. The body weight changes of untreated mice
(n=4) are indicated by black squares, and those of DAT-administered
mice (n=4) are indicated by gray circles. At least four mice were
tested for each experiment, and the error bars represent the
standard deviations. (C) Photograph of the liver, kidney, spleen,
heart and lung of DAT-treated and DAT-untreated BCBL1-xenografted
mice. Individual organs from DAT-untreated mice (Cont) and
DAT-treated mice (DAT) are on the left and right, respectively. (D)
Spleen weight of DAT-untreated or DAT-treated BCBL1-xenografted
SCID mice. The weight (Cont) of the spleen from DAT-untreated
BCBL1-injected mice is indicated by black bars and that (DAT) from
DAT-treated BCBL1-injected mice is indicated by gray bars. (E) The
weight of intraperitoneal tumor cells of DAT-untreated (Cont) or
DAT-treated (DAT) BCBL1-administered SCID mice. Tumor weights of
DAT-untreated mice (n=4) and DAT-treated mice are indicated by
black and gray bars, respectively. Ascites containing tumor cells
were collected from each mouse on day 21 after the first DAT
administration. Tumor cells were separated from ascites by
centrifugation, and tumor weight was measured. The wet weight of
the obtained precipitate was regarded as the tumor weight. (F) KSHV
genome of tumor cells in ascites from DAT-untreated (Cont) or
DAT-treated (DAT) BCBL1-administered mice. The KSHV genome copy
numbers from ascites cells were quantified by real-time PCR. (G)
KSHV genome of the spleens from DAT-untreated (Cont) or DAT-treated
(DAT) BCBL1-administered mice. The KSHV DNA copy numbers in the
spleen were quantified by real-time PCR. (H) LANA expression of
ascites cells from DAT-untreated BCBL1-administered mice. The
intraperitoneal tumor cells were collected from ascites of
DAT-untreated xenografted mice, and were fixed in methanol,
followed by incubation with anti-LANA antibody. (D–H) Ascites cells
and spleen were collected from BCBL1-xenografted SCID mice on day
21 after the first DAT or corn oil administration. Standard
deviations were determined by analysis of data from three
independent mice. *p<0.05 and **p<0.01
(t-test). |
Discussion
Our data showed that DAT exhibited the greatest
cytotoxicity against PEL among the allyl sulfides (DAS, DAD and
DAT) tested in this study (Fig.
1). DAT specifically inhibited the growth of PEL cell lines
compared with KSHV-uninfected cells both in vitro and in
vivo (Figs. 1C and 7). DAS, DAD and DAT have antitumor
effects, but DAS has been shown to have antioxidant and protective
effects against oxidative or chemical stress rather than cytotoxic
effects (31). On the other hand,
DAD and DAT show cytotoxic rather than cell-protective effects. In
particular, DAT was shown to induce apoptosis in cancer cells by
the generation of ROS and the dysregulation of signaling pathways,
including Bcl2-caspase-9, Erk, Jnk and Akt signaling (32,33).
By cleavage within its trisulfide bonds, DAT generates ROS, leading
to ER stress and apoptotic cell death (20–23,34).
In addition, DAT has been reported to suppress NF-κB and to induce
apoptosis, DAT reduced LPS-induced NF-κB transcriptional activation
in macrophages (35), and DAT
suppressed high-glucose-induced apoptosis by inhibiting NF-κB
signaling via decreases in nuclear translocation of NF-κB (36). Furthermore, DAT suppressed NF-κB
through an increase in IκBα in osteosarcoma cells, resulting in a
decrease in P-glycoprotein (37).
DAT suppresses dextran sodium sulfate-induced mouse colitis by
inducing STAT3 phosphorylation and suppression of IκBα
phosphorylation (24). However,
the detailed mechanism by which DAT inhibits NF-κB signaling has
not been determined. To our knowledge, this is the first report of
the detailed mechanism of dysregulation of NF-κB signaling by DAT.
Thus, DAT has the ability to interfere in cell signaling including
NF-κB, while constitutive and/or transient activation of NF-κB, Akt
and Erk signaling are necessary for PEL to enhance cell growth and
survival (4–11). These findings may be a reason why
DAT, rather than DAS and DAD, showed selective cytotoxic effect on
PEL cells.
The present study revealed a novel biological
function of DAT and a mechanism for DAT-mediated NF-κB suppression.
We propose a model in which TRAF6 downregulation by DAT results in
suppression of NF-κB signaling (Fig.
8). DAT induced the proteasomal degradation of TRAF6, and this
DAT-mediated TRAF6 downregulation suppressed IKKβ phosphorylation
(Fig. 5), resulting in inhibition
of the IKK complex. The suppression of the IKK complex by DAT
decreased the Ser32 and Ser36 phosphorylation of IκBα (Fig. 4C) followed by stabilization of IκBα
in a DAT-dependent manner (Fig.
3A). DAT induced stabilization of IκBα by these molecular
mechanisms, and then suppressed both the nuclear localization of
p65 and the transcriptional activity of NF-κB in PEL cells
(Fig. 3); in contrast, DAT did not
affect the stabilization of IκBα in KSHV-uninfected cells. It has
been suggested that TRAF6 induces the activation of IKK complex and
TAK1-TAB2 complex via K63-linked polyubiquitin (12–14).
TRAF6, an E3 ubiquitin ligase, catalyzes synthesis of K63-linked
polyubiquitination of IKKγ and TRAF6 itself (12). The K63-linked polyubiquitin chains
function as a scaffold to recruit the TAK1-TAB2 complex and IKK
complex through binding to TAB2 and IKKγ, respectively (13). Recruitment of the kinase complexes
facilitates phosphorylation of the catalytic subunit IKKβ by TAK1,
leading to activation of IKK complex. The activated IKK complex
induces phosphorylation of IκBα, which acts as a trigger for
K48-linked polyubiquitination by SCF-type E3 ubiquitin ligase
(38), and the subsequent
proteasomal degradation of IκBα. TRAF6 has the RING domain for
binding E2, and has been classified as a monomeric RING-type E3
ubiquitin ligase. TRAF6 interacts with Ubc13, which is one of the
E2s, forms a dimer with Uev1A (also called Mms2), and mediates the
K63-linked polyubiquitination of IKKγ and TRAF6 itself (13,39).
The functions of TRAF6 are not aimed at targeting proteins for
degradation, but to induce qualitative changes in polyubiquitinated
proteins, including stabilization (40), changing localization (41), and signal cascade activation
(13,14). It has been suggested that
K63-linked self-polyubiquitination of TRAF6 is important for signal
transduction from TRAF6 to downstream effectors, such as IKKγ or
TAK1-TAB2 complex. It is demonstrated that DAT induces the
proteasome-dependent degradation of TRAF6, leading to suppression
of NF-κB signaling. This suggests that DAT may induce malformation
or disorder of TRAF6-Ubc13-Uev1A complex, resulting in K48-linked
self-polyubiquitination of TRAF6 and subsequent TRAF6 degradation
by the 26S proteasome. It is possible that additional consequences
of DAT-induced TRAF6 degradation will become apparent upon further
characterization of additional participants in the TRAF6 complex in
the presence of DAT.
We propose that the PEL-specific antitumor activity
of DAT treatment is mainly due to the DAT-induced NF-κB inhibition.
Constitutive NF-κB activation is essential for the antiapoptosis
and growth of PEL cells (7–9),
consistent with our data, the NF-κB inhibitor BAY11-7082, decreased
the viability of PEL cells compared to KSHV-uninfected cells
(Fig. 4D), and other NF-κB
inhibitors also have PEL-specific cytotoxic effects (10,11).
In fact, KSHV targets NF-κB signaling to influence gene expression
and apoptosis. KSHV-encoded v-FLIP, K15, v-GPCR, K1 and K7 activate
NF-κB signaling to achieve antiapoptosis in KSHV-infected cells,
including PEL cells (4,6). Therefore, DAT-mediated
destabilization of TRAF6 induces stabilization of IκBα and the
subsequent inhibition of NF-κB signaling, which in turn may result
in apoptosis of PEL cells. NF-κB signaling is also required for
KSHV replication (10,11). DAT inhibited virus production at
low concentrations, such as 0.2 and 1.0 μM (Fig. 6A), which do not influence
proliferation of PEL cells (Fig.
1). DAT administration suppressed the ascites in the peritoneal
cavity and the development of KSHV-infected cells in ascites of
PEL-xenografted SCID mice (Fig.
7). DAT significantly inhibited the growth and infiltration of
PEL cells in vivo and in vitro. Taken together, our
data suggest that DAT may serve as a new lead compound for the
development of novel drugs against not only PEL but also KSHV
infection.
Acknowledgements
This study was supported by the MEXT-Supported
Program for the Strategic Research Foundation at Private
Universities (S1311035) and JSPS KAKENHI (24590103).
Abbreviations:
|
DAD
|
diallyl disulfide
|
|
DAS
|
diallyl sulfide
|
|
DAT
|
diallyl trisulfide
|
|
HHV-8
|
human herpes virus-8
|
|
IκB
|
inhibitor of NF-κB
|
|
IKK
|
IκB kinase
|
|
KSHV
|
Kaposi's sarcoma-associated
herpesvirus
|
|
LANA
|
latency associated nuclear antigen
|
|
PEL
|
primary effusion lymphoma
|
|
TAB
|
TAK1-binding protein
|
|
TRAF6
|
TNF-α receptor-associated factor 6
|
|
TAK1
|
TGF-β-activated kinase 1
|
References
|
1
|
Russo JJ, Bohenzky RA, Chien MC, Chen J,
Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, et
al: Nucleotide sequence of the Kaposi sarcoma-associated
herpesvirus (HHV8). Proc Natl Acad Sci USA. 93:14862–14867. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: A
distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.PubMed/NCBI
|
|
3
|
Chang Y, Cesarman E, Pessin MS, Lee F,
Culpepper J, Knowles DM and Moore PS: Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma.
Science. 266:1865–1869. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Järviluoma A and Ojala PM: Cell signaling
pathways engaged by KSHV. Biochim Biophys Acta. 1766:140–158.
2006.PubMed/NCBI
|
|
5
|
Fujimuro M, Wu FY, ApRhys C, Kajumbula H,
Young DB, Hayward GS and Hayward SD: A novel viral mechanism for
dysregulation of beta-catenin in Kaposi's sarcoma-associated
herpesvirus latency. Nat Med. 9:300–306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ashizawa A, Higashi C, Masuda K, Ohga R,
Taira T and Fujimuro M: The ubiquitin system and Kaposi's
sarcoma-associated herpesvirus. Front Microbiol. 3:662012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chaudhary PM, Jasmin A, Eby MT and Hood L:
Modulation of the NF-κB pathway by virally encoded death effector
domains-containing proteins. Oncogene. 18:5738–5746. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Keller SA, Schattner EJ and Cesarman E:
Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary
effusion lymphoma cells. Blood. 96:2537–2542. 2000.PubMed/NCBI
|
|
9
|
Keller SA, Hernandez-Hopkins D, Vider J,
Ponomarev V, Hyjek E, Schattner EJ and Cesarman E: NF-kappaB is
essential for the progression of KSHV- and EBV-infected lymphomas
in vivo. Blood. 107:3295–3302. 2006. View Article : Google Scholar
|
|
10
|
Saji C, Higashi C, Niinaka Y, Yamada K,
Noguchi K and Fujimuro M: Proteasome inhibitors induce apoptosis
and reduce viral replication in primary effusion lymphoma cells.
Biochem Biophys Res Commun. 415:573–578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Higashi C, Saji C, Yamada K, Kagawa H,
Ohga R, Taira T and Fujimuro M: The effects of heat shock protein
90 inhibitors on apoptosis and viral replication in primary
effusion lymphoma cells. Biol Pharm Bull. 35:725–730. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang G, Gao Y, Li L, Jin G, Cai Z, Chao JI
and Lin HK: K63-linked ubiquitination in kinase activation and
cancer. Front Oncol. 2:52012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen ZJ: Ubiquitination in signaling to
and activation of IKK. Immunol Rev. 246:95–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Skaug B, Jiang X and Chen ZJ: The role of
ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem.
78:769–796. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calvo-Gómez O, Morales-López J and López
MG: Solid-phase microextraction-gas chromatographic-mass
spectrometric analysis of garlic oil obtained by hydrodistillation.
J Chromatogr A. 1036:91–93. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lawson LD, Wang ZJ and Hughes BG:
Identification and HPLC quantitation of the sulfides and
dialk(en)yl thiosulfinates in commercial garlic products. Planta
Med. 57:363–370. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yi L and Su Q: Molecular mechanisms for
the anti-cancer effects of diallyl disulfide. Food Chem Toxicol.
57:362–370. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang HC, Pao J, Lin SY and Sheen LY:
Molecular mechanisms of garlic-derived allyl sulfides in the
inhibition of skin cancer progression. Ann N Y Acad Sci.
1271:44–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
You S, Nakanishi E, Kuwata H, Chen J,
Nakasone Y, He X, He J, Liu X, Zhang S, Zhang B, et al: Inhibitory
effects and molecular mechanisms of garlic organosulfur compounds
on the production of inflammatory mediators. Mol Nutr Food Res.
57:2049–2060. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Das A, Banik NL and Ray SK: Garlic
compounds generate reactive oxygen species leading to activation of
stress kinases and cysteine proteases for apoptosis in human
glioblastoma T98G and U87MG cells. Cancer. 110:1083–1095. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chandra-Kuntal K, Lee J and Singh SV:
Critical role for reactive oxygen species in apoptosis induction
and cell migration inhibition by diallyl trisulfide, a cancer
chemopreventive component of garlic. Breast Cancer Res Treat.
138:69–79. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang HC, Hsieh SC, Yang JH, Lin SY and
Sheen LY: Diallyl trisulfide induces apoptosis of human basal cell
carcinoma cells via endoplasmic reticulum stress and the
mitochondrial pathway. Nutr Cancer. 64:770–780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Filomeni G, Rotilio G and Ciriolo MR:
Molecular transduction mechanisms of the redox network underlying
the antiproliferative effects of allyl compounds from garlic. J
Nutr. 138:2053–2057. 2008.PubMed/NCBI
|
|
24
|
Lee HJ, Lee HG, Choi KS, Surh YJ and Na
HK: Diallyl trisulfide suppresses dextran sodium sulfate-induced
mouse colitis: NF-κB and STAT3 as potential targets. Biochem
Biophys Res Commun. 437:267–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin DY, Kim GY, Hwang HJ, Kim WJ and Choi
YH: Diallyl trisulfide-induced apoptosis of bladder cancer cells is
caspase-dependent and regulated by PI3K/Akt and JNK pathways.
Environ Toxicol Pharmacol. 37:74–83. 2014. View Article : Google Scholar
|
|
26
|
Okada S, Goto H and Yotsumoto M: Current
status of treatment for primary effusion lymphoma. Intractable Rare
Dis Res. 3:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wakao K, Watanabe T, Takadama T, Ui S,
Shigemi Z, Kagawa H, Higashi C, Ohga R, Taira T and Fujimuro M:
Sangivamycin induces apoptosis by suppressing Erk signaling in
primary effusion lymphoma cells. Biochem Biophys Res Commun.
444:135–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fujimuro M, Liu J, Zhu J, Yokosawa H and
Hayward SD: Regulation of the interaction between glycogen synthase
kinase 3 and the Kaposi's sarcoma-associated herpesvirus
latency-associated nuclear antigen. J Virol. 79:10429–10441. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fujimuro M, Sawada H and Yokosawa H:
Production and characterization of monoclonal antibodies specific
to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett.
349:173–180. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen C and Okayama H: High-efficiency
transformation of mammalian cells by plasmid DNA. Mol Cell Biol.
7:2745–2752. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Milner JA: A historical perspective on
garlic and cancer. J Nutr. 131(Suppl 3): 1027S–1031S.
2001.PubMed/NCBI
|
|
32
|
Xiao D, Choi S, Johnson DE, Vogel VG,
Johnson CS, Trump DL, Lee YJ and Singh SV: Diallyl
trisulfide-induced apoptosis in human prostate cancer cells
involves c-Jun N-terminal kinase and extracellular-signal regulated
kinase-mediated phosphorylation of Bcl-2. Oncogene. 23:5594–5606.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Antosiewicz J, Herman-Antosiewicz A,
Marynowski SW and Singh SV: c-Jun NH(2)-terminal kinase signaling
axis regulates diallyl trisulfide-induced generation of reactive
oxygen species and cell cycle arrest in human prostate cancer
cells. Cancer Res. 66:5379–5386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murai M, Inoue T, Suzuki-Karasaki M,
Ochiai T, Ra C, Nishida S and Suzuki-Karasaki Y: Diallyl trisulfide
sensitizes human melanoma cells to TRAIL-induced cell death by
promoting endoplasmic reticulum-mediated apoptosis. Int J Oncol.
41:2029–2037. 2012.PubMed/NCBI
|
|
35
|
Liu KL, Chen HW, Wang RY, Lei YP, Sheen LY
and Lii CK: DATS reduces LPS-induced iNOS expression, NO
production, oxidative stress, and NF-kappaB activation in RAW 264.7
macrophages. J Agric Food Chem. 54:3472–3478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kuo WW, Wang WJ, Tsai CY, Way CL, Hsu HH
and Chen LM: Diallyl trisufide (DATS) suppresses high
glucose-induced cardiomyocyte apoptosis by inhibiting JNK/NFκB
signaling via attenuating ROS generation. Int J Cardiol.
168:270–280. 2013. View Article : Google Scholar
|
|
37
|
Wang Z, Xia Q, Cui J, Diao Y and Li J:
Reversion of P-glycoprotein-mediated multidrug resistance by
diallyl trisulfide in a human osteosarcoma cell line. Oncol Rep.
31:2720–2726. 2014.PubMed/NCBI
|
|
38
|
Nakayama KI and Nakayama K: Ubiquitin
ligases: Cell-cycle control and cancer. Nat Rev Cancer. 6:369–381.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hershko A and Ciechanover A: The ubiquitin
system for protein degradation. Annu Rev Biochem. 61:761–807. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choi YB and Harhaj EW: HTLV-1 tax
stabilizes MCL-1 via TRAF6-dependent K63-linked polyubiquitination
to promote cell survival and transformation. PLoS Pathog.
10:e10044582014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang WL, Wang J, Chan CH, Lee SW, Campos
AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al: The E3
ligase TRAF6 regulates Akt ubiquitination and activation. Science.
325:1134–1138. 2009. View Article : Google Scholar : PubMed/NCBI
|