Introduction
The flavone apigenin (4′,5,7,-trihydroxyflavone), is
a plant secondary metabolite that occurs widely in numerous fruits.
It is a potent anticancer drug that can suppress cancer growth,
angiogenesis, and metastasis in multiple types of cancer (1). Mechanistically, several studies have
shown that apigenin suppresses the activation of nuclear
transcription factor-κB (NF-κB), phosphoinositide 3-kinase (PI3K),
epidermal growth factor receptor (EGFR), hypoxia-inducible
factor-1α (HIF-1α), vascular endothelial growth factor (VEGF),
cyclin D1, and vascular cell adhesion molecule 1 (VCAM-1), which
play critical roles in the regulation of cell growth, cell cycle,
angiogenesis, metastasis, and apoptosis (2). In addition, several studies have
shown that apigenin causes accumulation of intracellular reactive
oxygen species (ROS) and oxidative stress through the depletion of
cellular glutathione (GSH), an important factor in redox balance
(3–6). Nevertheless, the precise molecular
mechanism by which decreased cellular GSH levels and increased
levels of ROS relate to apigenin-mediated apoptosis is not fully
understood.
Cancer cells use large amounts of glucose and
glutamine to meet the increased energetic and anabolic demands
associated with unlimited cell growth and survival (7–9).
Increased glucose utilization in cancer cells is achieved primarily
by upregulation of the expression of glucose transporter 1 (GLUT1),
which is widely expressed in many tumors, including hepatic,
pancreatic, breast, brain, lung, colorectal, and cervical cancers
(10). Indeed, several reports
have shown that GLUT1 inhibitors, such as fasentin, phloretin, and
WZB117, suppress growth and induce cell death in cancer cells
(11–13). Upregulation of GLUT1 expression is
closely associated with chemoresistance, tumor aggressiveness, and
decreased survival among patients (14,15).
Therefore, downregulation of GLUT1 expression could be a promising
therapeutic strategy for cancer treatment.
The pentose phosphate pathway (PPP), which is
required for biosynthesis of ribonucleotides and NADPH, can be
initiated by glucose-6-phosphate generated by hexokinase as an
initial key enzyme in glucose metabolism (16). As a cellular reducing power, NADPH
can remove ROS through regulation of the GSH and thioredoxin (TRX)
systems that directly scavenge ROS and repair ROS-induced cellular
damage (7). Thus, inhibition of
NADPH production has been proposed as a clinical intervention for
cancer treatment. Indeed, preclinical studies have shown that
inhibition of glucose-6-phosphate dehydrogenase (G6PD), an enzyme
that initiates the PPP, is sufficient to decrease cell growth in
leukemia, glioblastoma, and lung cancer cells (17).
Glutamine is a critical energy source required to
maintain cell growth and survival. Growing cancer cells are
commonly dependent upon a supply of glutamine to support the
tricarboxylic acid (TCA) cycle. Mechanistically, glutamine can be
converted to glutamate and α-ketoglutarate (α-KG) via the enzymes
glutaminase (GLS) and glutamate dehydrogenase (GDH) (18). Previous studies have shown that
inhibition of glutamine metabolism by GLS inhibitors, including
6-diazo-5-oxo-L-norleucine (DON),
Bis-2-[5-phenylacetamido-1,2,4-thiadiazol-2-yl] ethyl sulfide
(BPTES), and compound 968, or genetic targeting of GLS or GDH using
small interfering RNA (siRNA), can suppress cell viability and
increase apoptosis in cancer cells. This suggests that inhibition
of glutamine utilization might cause cancer cells to become more
sensitive to the effects of conventional anticancer drugs (18–20).
However, there is no available information addressing synergistic
effects of apigenin on targeting cancer specific metabolism, such
as glutamine metabolism.
In this study, we examined whether apigenin: i)
inhibits GLUT1 expression and glucose utilization, thereby
suppressing cell growth and inducing apoptosis; ii) downregulates
NADPH and GSH production by inhibition of glucose metabolism,
thereby resulting in increased oxidative stress; and iii)
sensitizes lung cancer cells to inhibition of glutamine
utilization, thereby causing apoptosis and growth retardation.
Materials and methods
Reagents and antibodies
N-acetyl-L-cysteine (NAC), apigenin,
2′-7′-dichlorodihydrofluorescein diacetate (DCF-DA) and compound
968 were purchased from Sigma-Aldrich (St. Louis, MO, USA) or
Millipore (Billerica, MA, USA). Antibodies recognizing cleaved
caspase-9, cleaved caspase-3, PARP, and HK1 (catalog nos. 9502,
9661, 9546, and 2024, respectively) were purchased from Cell
Signaling Technology (Danvers, MA, USA). GLUT1 (sc-7903), GLS1
(156876) and β-tubulin (sc-9104) were purchased from Santa Cruz
Biotechnology (Dallas, TX, USA) and Abcam (Cambridge, MA, USA).
Cell culture, plasmids, shRNA, and
generation of stable cell lines
Lung cancer cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum and 25 mM glucose. The lentiviral vector pLenti-GIII-CMV-
SLC2A1, expressing human GLUT1 was obtained from Applied Biological
Materials Inc. (Richmond, BC, Canada). A short hairpin RNA
construct targeting GLS1 was purchased from Sigma-Aldrich. The
hairpin sequence was: 5′-CCGGG CCCTGAAGCAGTTCGAAATACTCGAGTATTTCGAAC
TGCTTCAGGGCTTTTTG-3′. Lentiviral particles were produced in HEK293T
cells, harvested 48 h after transfection, and then used to infect
lung cancer cells. Infected lung cancer cells were selected by
growth on media containing 2 μg/ml of puromycin (Invitrogen,
Carlsbad, CA, USA) for 6 days.
Western blotting
Cells were lysed in buffer containing 1% IGEPAL, 150
mM NaCl, 50 mM Tris-HCl (pH 7.9), 10 mM NaF, 0.1 mM EDTA, and
protease inhibitor cocktail. Protein samples were separated by
SDS-PAGE, and the separated proteins were then transferred to a
PVDF membrane (Millipore). Membranes were incubated in the presence
of primary antibodies (1:1,000) at 4°C overnight, and
HRP-conjugated secondary antibodies (1:10,000) were incubated for 1
h at room temperature. Proteins were visualized using an enhanced
chemiluminescence (ECL) Prime kit (GE Healthcare, Pittsburgh, PA,
USA).
ROS measurement
To measure ROS levels, cells were incubated with
Hank's balanced salt solution (HBSS), containing 30 μM DCF-DA, for
30 min. After incubation, cells were detached and resuspended in
phosphate-buffered saline (PBS) in preparation for
fluorescence-activated cell sorting (FACS) analysis.
RNA isolation and RT-qPCR
Total RNA was extracted from cells using TRIzol
(Invitrogen), and 2 μg of total RNA was used for cDNA synthesis
using a high capacity cDNA reverse transcription kit (Applied
Biosystems). Quantitative PCR was performed using SYBR Green PCR
Master Mix (Applied Biosystems). The sequences of the PCR primers
(5′-3′) were: GAGAGTTTCATCGGAGAGCC and CAGCGAGAATCGG CTACAG for
HK1; GGCATTGATGACTCCAGTGTT and ATGGAGCCCAGCAGCAA for GLUT1;
CTTGGGACAG CAGCCTTAAT and CAAGCTGGACGTTAAAGGGA for PGK1;
AGCCCTTTCTCCATCTCCTT and AACCATGAC CAAGTGCAGAA for HK2;
TACAGGCACAGTCGCAG AGT and CACTTCCTGGATGCTTGCTG for ALDOA;
CCTGGCATGGATCTTGAGAA and TACGTTCACCT CGGTGTCTG for ENO1.
Glucose consumption, ATP, and lactate
production assays
As previously described (9), lactate and glucose levels in cultured
media were measured using lactate colorimetric assay kit and
glucose colorimetric assay kit, respectively (BioVision, USA).
Briefly, cells were cultured in DMEM without phenol red for 24 h,
and then cultured media were mixed with assay solution. To measure
intracellular ATP levels, cells were cultured in 6-well plates, and
cell lysates were mixed with reaction buffer. ATP levels in the
cell lysates were then determined using a
luciferin-luciferase-based assay kit (Invitrogen), used according
to the manufacturer's instructions. All values were normalized
relative to cellular protein concentration.
Glutathione and NADPH assays
To prepare samples for determination of glutathione
levels, cells were cultured in 6-well tissue culture dishes, and
then cell lysates were obtained in extraction buffer without
dithiothreitol (DTT) or β-mercaptoethanol. The levels of reduced
glutathione (GSH) and oxidized glutathione (GSSG) were measured
using a Glutathione Colorimetric Detection kit (BioVision) and an
OxiSelect Glutathione Assay kit (Cell Biolabs), respectively, as
previously described (21). The
levels of NADPH in cultured cells were measured using a NADPH
quantitation kit (BioVision). Briefly, cell lysates were extracted
in NADPH extraction buffer, and then NADPH levels were determined
by measurement of absorption at a wavelength of 450 nm.
Clonogenic cell survival assays
To measure clonogenic cell survival,
1×104 lung cancer cells were seeded in 6-well tissue
culture dishes, and incubated for 7 days. After incubation, cells
were fixed in 10% paraformaldehyde, and then stained with crystal
violet for 10 min.
Apoptosis assays
A kit utilizing Annexin V-FITC and propidium iodide
(BD Biosciences, San Jose, CA, USA) was used to quantify the
numbers of apoptotic cells. Briefly, cells were detached from the
culture dishes, washed with cold PBS, and then incubated with
anti-Annexin V-FITC antibody and propidium iodide. Apoptotic cell
numbers were determined by FACS analysis.
Statistical analysis
All data were analyzed using the unpaired Student's
t-test for two experimental comparisons and one-way ANOVA with
Tukey post-test for multiple comparisons. Data are represented as
means ± standard deviations (SD). A p-value <0.05 was considered
statistically significant.
Results
Apigenin reduces GLUT1 expression
levels
Because it has been reported that apigenin exerts an
anticancer effect through suppression of GLUT1 expression (22–25),
we examined whether apigenin also downregulates gene expression
related to glycolysis, including GLUT1, HK1, PGK1, HK2, ALDOA, and
ENO1. The results were consistent with those of previous reports
(22–25) that apigenin significantly decreased
GLUT1 mRNA (Fig. 1A) and protein
(Fig. 1B) levels, but did not
decrease the levels of other glycolytic enzymes in H1299 or H460
lung carcinoma cells. In addition, GLUT1 mRNA levels were also
decreased by apigenin treatment in other cancer cell lines,
including A549 (lung carcinoma), H460 (lung carcinoma), H2030 (lung
adenocarcinoma), HCT116 (colorectal carcinoma), SW480 (colorectal
adenocarcinoma), and A375 (melanoma) (Fig. 1C). To elucidate whether apigenin
suppresses GLUT1 expression in mice, we measured GLUT1 and GLUT4
expression levels in skeletal muscle, white adipose tissue, the
heart, brain, liver, lung, and pancreas obtained from
apigenin-treated mice. Fig. 1D
shows that apigenin significantly suppressed GLUT1 but not GLUT4
expression in the brain, liver, lung, and pancreas. These results
indicate that apigenin decreases GLUT1 mRNA and protein levels in
various human cancer cell lines, and in mice. All experiments were
performed according to the guidelines of the Animal Care and Use
Committee, Konkuk University, Korea.
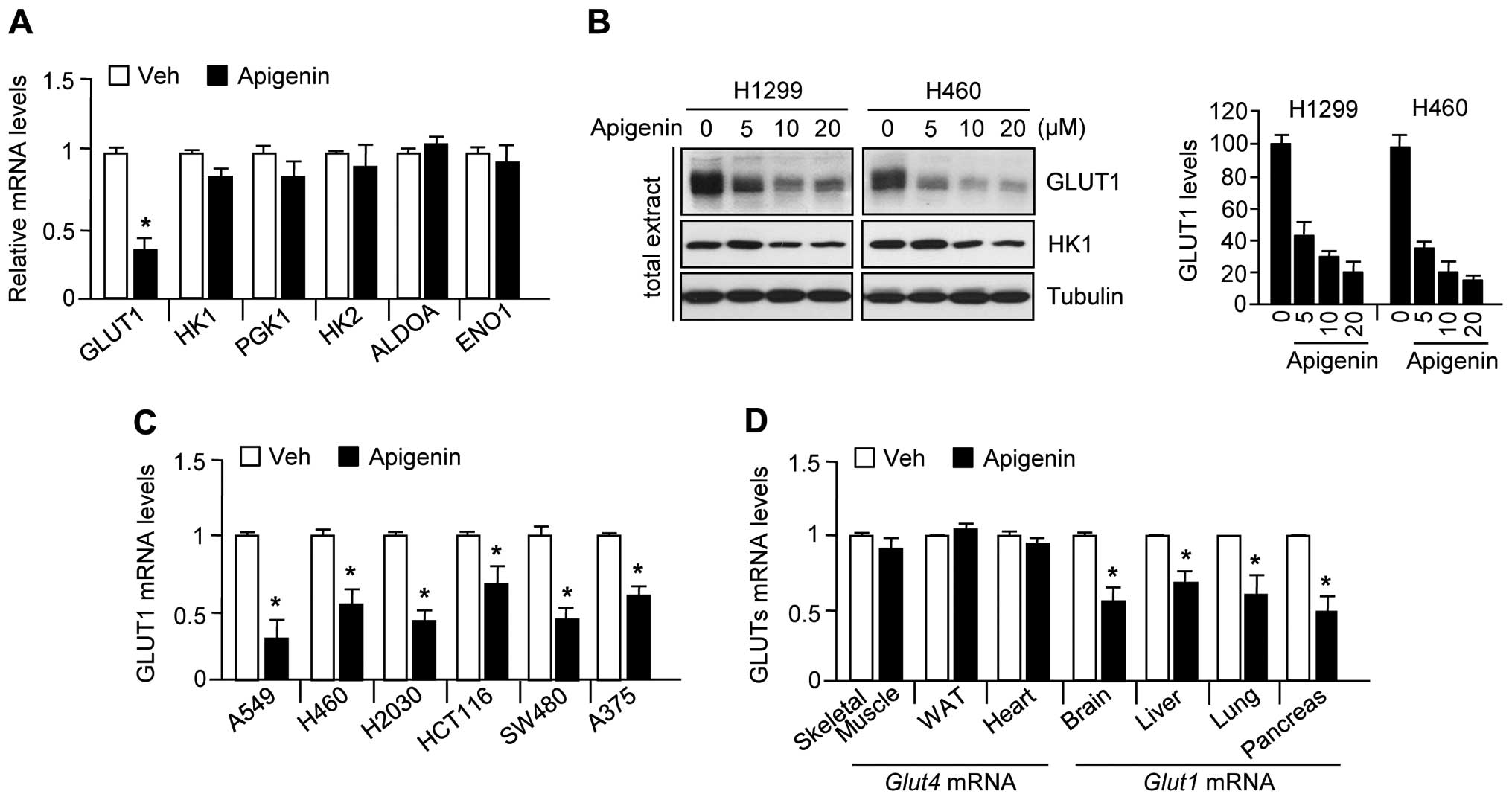 | Figure 1Apigenin decreases GLUT1 mRNA and
protein levels in several cancer cell lines. (A) H1299 (lung
carcinoma) cells were incubated in the absence or presence of 10 μM
apigenin for 24 h, and then glycolytic gene expression levels were
assessed by quantitative RT-PCR. Values represent mean ± SD of
three independent experiments performed in triplicate;
*p<0.05. (B) GLUT1 protein levels were determined by
immunoblotting (n=3) and quantified by ImageJ. (C) GLUT1 mRNA
levels were measured by quantitative RT-PCR in A549 (lung
carcinoma), H460 (lung carcinoma), H2030 (lung adenocarcinoma),
HCT116 (colorectal carcinoma), SW480 (colorectal adenocarcinoma),
and A375 (melanoma). (D) Mice were injected daily with apigenin (1
mg/kg) or DMSO for 10 days. GLUT1 and GLUT4 mRNA levels in the
liver, lung, brain, pancreas, skeletal muscle, white adipose, and
heart were measured by quantitative RT-PCR. Values represent mean ±
SD (n=6); *p<0.05. |
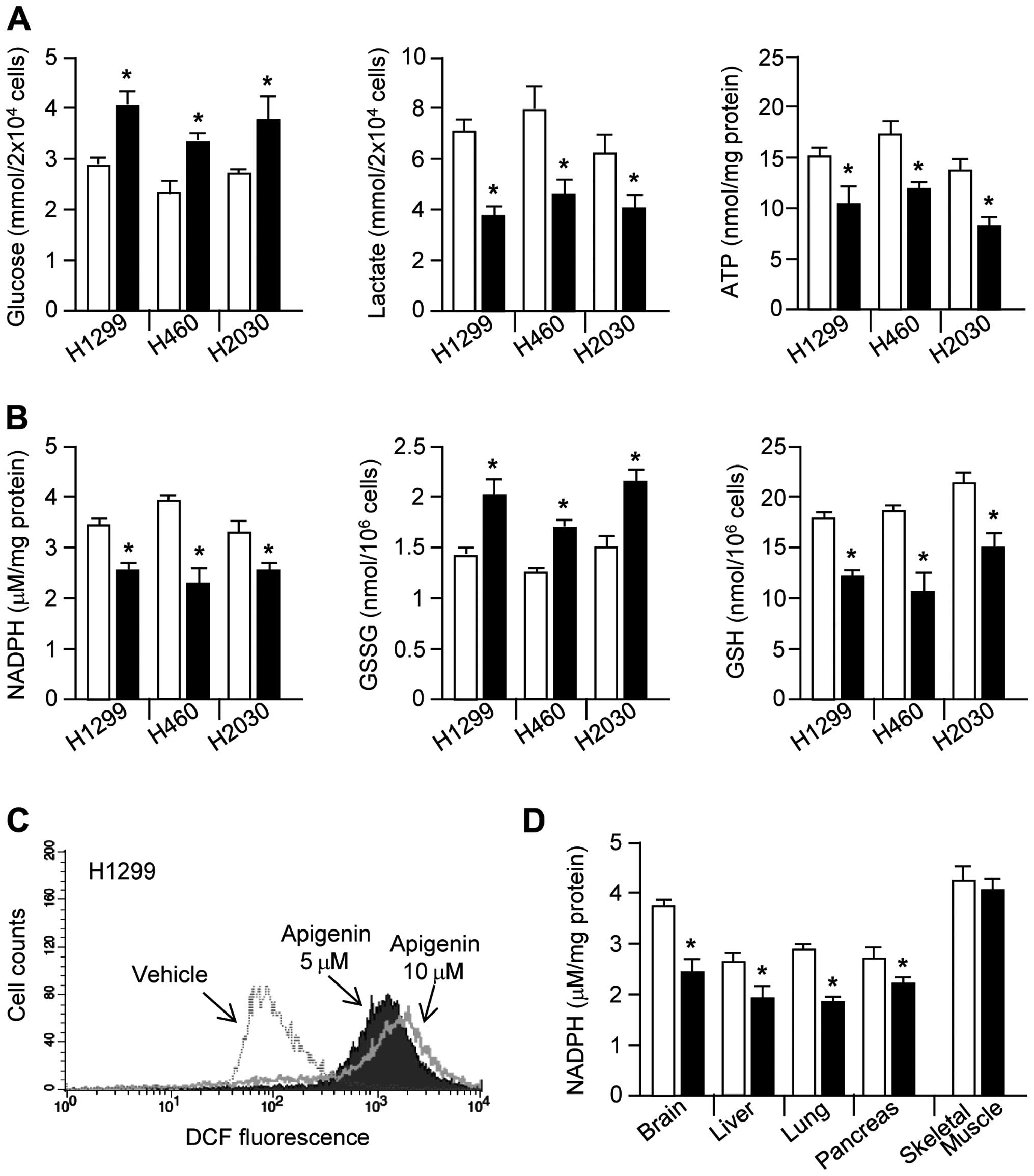
Apigenin causes cellular metabolic and
oxidative stress
Cancer cells use large amounts of glucose to
generate glycolytic intermediates and end products, including
lactate, ATP, nucleotides, lipids, amino acids, and NADPH, all of
which contribute to rapid cell proliferation (8). Therefore, we investigated whether
apigenin can block glucose utilization in cancer cells.
Interestingly, glucose consumption, lactate production, and ATP
production were all strongly decreased by apigenin treatment
(Fig. 2A). Glucose-6-phosphate,
which is produced from glucose, can be shunted into the oxidative
branch of the PPP by glucose-6-phosphate dehydrogenase, thereby
generating NADPH and promoting redox homeostasis (26,27).
Here, we demonstrated that NADPH generation by the oxidative branch
of the PPP is decreased by apigenin treatment. Fig. 2B shows that, in apigenin-treated
lung cancer cells, NADPH and GSH levels were significantly
decreased, whereas GSSG levels were increased. Since NADPH
maintains redox homeostasis through generation of GSH, which is
necessary for the elimination of hydrogen peroxide
(H2O2) by glutathione peroxidase (GPx)
(27), we measured intracellular
ROS levels. As we had hypothesized, intracellular ROS levels were
strongly increased in apigenin-treated H1299 cells (Fig. 2C). Fig. 2D shows significantly decreased
NADPH levels in the brain, liver, lung, and pancreatic tissues
derived from apigenin-treated mice. These results suggest that
apigenin induces oxidative stress through destruction of glucose
utilization-mediated redox homeostasis.
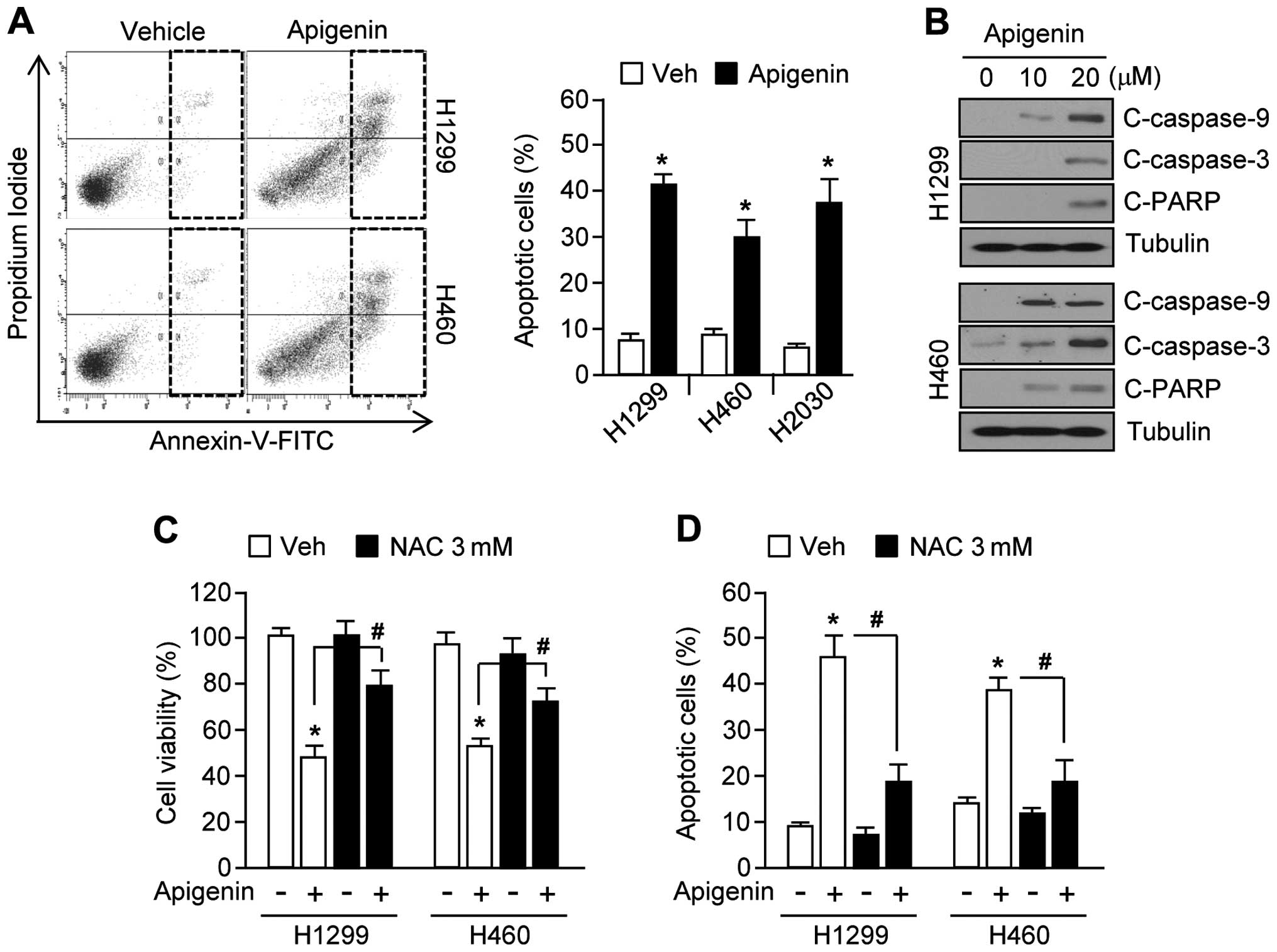
Apigenin causes oxidative stress leading
to apoptosis
Because apoptotic signal transduction cascades
involving caspase-9, -3 and PARP cleavage can be activated by
increased ROS levels (9,28), we examined whether apigenin could
likewise increase apoptosis through this pathway. Fig. 3A shows that apoptotic cell numbers
were increased by ~30% in apigenin-treated H1299, H460, and H2030
lung cancer cells. Furthermore, in H1299 and H460 cells, apigenin
increased caspase-9, -3 and PARP cleavage in a dose-dependent
manner (Fig. 3B). In addition,
apigenin-induced apoptosis may be mediated by elevated ROS levels
in H1299 and H460 cells, because treatment with the antioxidant,
N-acetyl-L-cysteine (NAC), significantly rescued cell
viability (Fig. 3C), and
suppressed cell apoptosis (Fig.
3D). Taken together, these results show that accumulation of
ROS mediates apigenin-induced apoptosis.
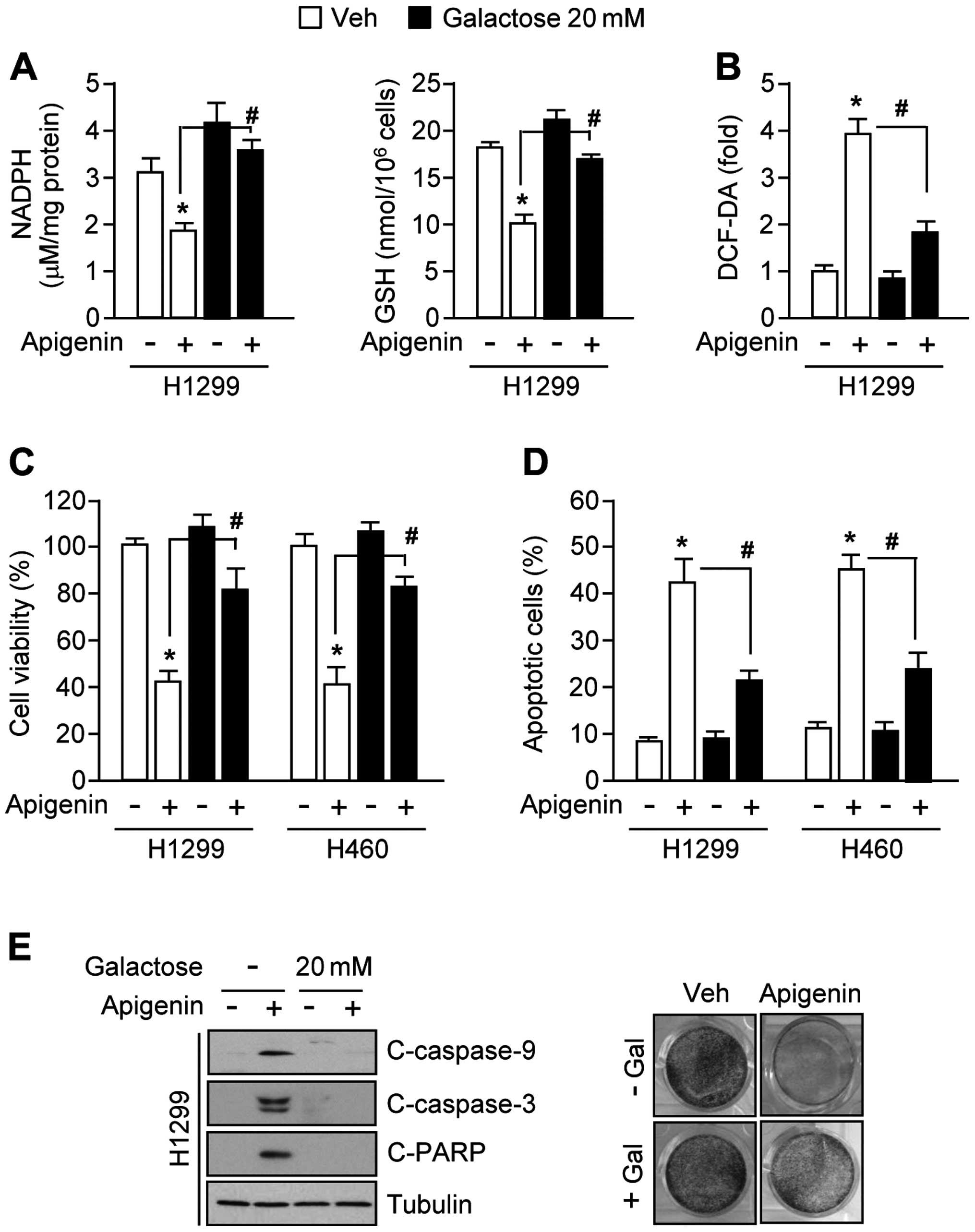
Galactose supplementation confers
resistance to apigenin-induced apoptosis by re-activating
antioxidant capacity
Because galactose supports NADPH production and
cancer cell proliferation largely through its metabolism in the
pentose phosphate pathway (26,29),
we examined whether galactose supplementation could attenuate
apigenin-mediated oxidative stress. Interestingly, the decreased
levels of NADPH and GSH in apigenin-treated H1299 cells were
strongly reversed in the presence of 20 mM galactose (Fig. 4A). Moreover, galactose decreased
ROS levels by ~50% (Fig. 4B).
Fig. 4C shows that galactose
increased cell viability by ~40% in apigenin-treated H1299 and H460
cells. Consistent with these results, the numbers of apoptotic
cells were also markedly decreased by ~20% in the presence of
apigenin with galactose supplemented cells (Fig. 4D). Fig. 4E shows that apigenin-mediated
activation of caspase-9, -3 and PARP apoptotic pathways (left
panel) and decreased clonogenic cell survival (right panel) were
completely blocked by 20 mM galactose in H1299 lung cancer cells.
Together, these results indicate that galactose confers resistance
to apigenin-mediated apoptosis by activating NADPH production
through the pentose phosphate pathway.
Cancer cells expressing high levels of
GLUT1 are resistant to apigenin-induced apoptosis through metabolic
compensation of glucose utilization
Because apigenin induces cancer cell death by
suppressing GLUT1 expression and glucose utilization in multiple
types of cancer cell lines, we examined whether
GLUT1-overexpressing cancer cells could confer resistance to
apigenin-induced apoptosis. For this purpose, we generated H1299
cell stably expressing a GLUT1 lentiviral vector, or an empty
vector (Fig. 5A). Fig. 5B shows that, even in the presence
of apigenin, glucose consumption increased dramatically in
GLUT1-overexpressing H1299 cells compared to cells harboring the
empty vector. Furthermore, GLUT1 overexpression produced high
levels of lactate in apigenin-treated H1299 cells compared to
levels of lactate in cells carrying the empty vector. To determine
whether GLUT1 overexpression could block apigenin-mediated
suppression of NADPH and GSH production, we measured intracellular
NADPH and GSSG levels in GLUT1-overexpressing H1299 cells in the
presence or absence of apigenin. Interestingly, the effects of
apigenin on NADPH and GSH levels were significantly rescued by
GLUT1 overexpression (Fig. 5C).
Furthermore, GLUT1 overexpression attenuated the effect of apigenin
on ROS, resulting in a clear decrease in ROS levels of ~50%
(Fig. 5D). Because increased
oxidative stress is a critical step for apigenin-mediated
apoptosis, we examined whether GLUT1-overexpressing cells were
resistant to apigenin-mediated cell death. Fig. 5E shows that 50 μM apigenin was
required to decrease cell viability in GLUT1-overexpressing cells,
while 10 μM apigenin was sufficient to decrease cell viability in
cells harboring the empty vector. Fig.
5F shows that 30 μM apigenin induced 20% of the cells to
undergo apoptosis in GLUT1-overexpressing H1299 cells. In contrast,
30 μM apigenin induced 50% of cells harboring the empty vector to
undergo apoptosis. Furthermore, GLUT1 overexpression dramatically
reversed the apigenin-mediated suppression of clonogenic cell
survival (Fig. 5G, left panel) and
attenuated the activation of caspase-9 and caspase-3 in H1299 cells
(Fig. 5G, right panel). These
results suggest that GLUT1 overexpression causes resistance to
apigenin-mediated apoptosis through suppression of metabolic and
oxidative stress.
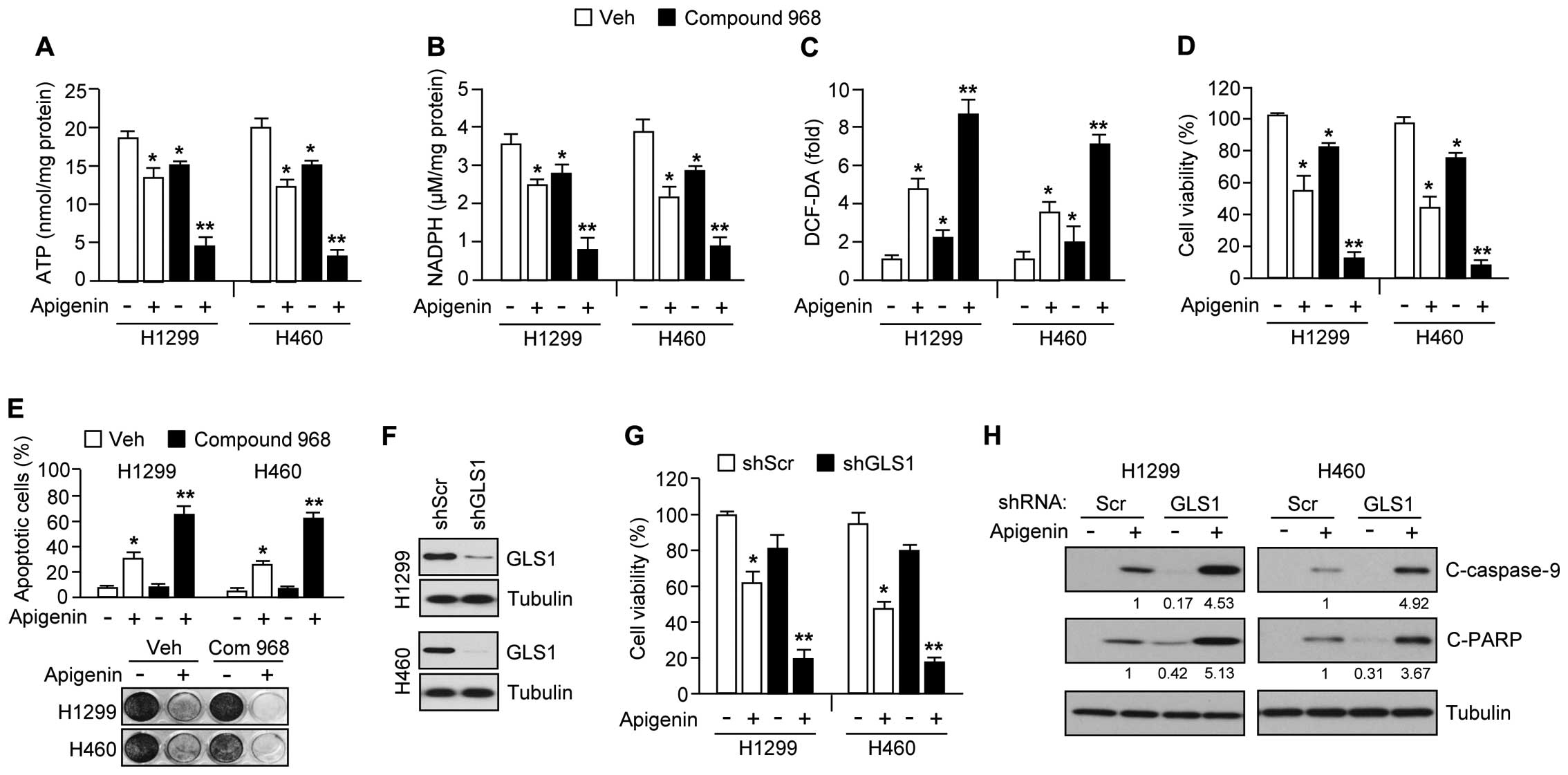
Inhibition of glutamine utilization
sensitizes apigenin-induced apoptosis through exacerbation of
metabolic stress
In cancer cells, glutamine is a nutrient critical
for promotion of tumor growth and survival (30,31).
Thus, inhibitors targeting glutamine utilization have shown
potential as anticancer drugs (18–20).
We therefore hypothesized that inhibition of glutamine utilization
might sensitize cancer cells to apigenin-mediated cell death, and
that the mechanism would involve suppression of the major pathways
of glucose and glutamine metabolism required for tumor growth and
survival. In support of this idea, Fig. 6A shows that compound 968, a GLS
inhibitor, synergized with apigenin, resulting in an ~75% reduction
of ATP levels in H1299 and H460 cells. Because glutamine
utilization supports NADPH production needed for redox control
(31), we additionally examined
whether combination treatment of apigenin and compound 968 is
sufficient to increase severe oxidative stress by decreasing NADPH
and increasing intracellular ROS levels in cancer cells. Here, we
found that compound 968 hugely decreased NADPH (Fig. 6B) and increased intracellular ROS
levels (Fig. 6C) in the presence
of apigenin. Furthermore, apigenin-treated cells, compound 968
exacerbated the reduction in cell viability (Fig. 6D), increased the induction of
apoptosis (Fig. 6E, upper panel),
and reduced clonogenic cell survival (Fig. 6E, bottom panel). In addition, we
used a short hairpin RNA (shRNA) to knock down GLS1 expression by
>70% in H1299 and H460 cell (Fig.
6F). Fig. 6G shows that
apigenin caused a large decrease in cell viability of ~80% in GLS1
knocked-down H1299 and H460 cells. Consistent with this, caspase-9
and PARP were more strongly activated in these cells (Fig. 6H), suggesting that decreased GLS1
expression or activity would make cancer cells more susceptible to
apigenin-mediated apoptosis. Taken together, these results indicate
that inhibition of glutamine utilization should sensitize lung
cancer cells to apigenin-mediated apoptosis.
Discussion
In recent years, apigenin, which is abundant in
common fruits and vegetables, has shown considerable promise for
development as an anticancer and chemopreventive agent (2,3,6). In
fact, several recent reports have shown that apigenin is capable of
selectively suppressing growth and inducing apoptosis in cancer
cells by affecting the activities of protein tyrosine kinases,
including epidermal growth factor receptor (EGFR), Src tyrosine
kinase, phosphatidylinositol 3-kinase (PI3K), mitogen-activated
protein kinase (MAPK), and hypoxia-inducible factor-1α (HIF-1Kα)
(32–36). It has been reported that apigenin
suppresses glucose uptake by a mechanism involving decreased GLUT1
expression, leading to increased apoptosis and chemosensitivity in
pancreatic carcinoma, laryngeal carcinoma, and head and neck cancer
(22–25). However, there is no previous
evidence to indicate that a combination of apigenin with other
inhibitors of cancer metabolism would show a synergistic effect in
the suppression of cancer growth and survival. In this study, we
observed that downregulation of GLUT1 by apigenin led to
suppression of glucose utilization through decreased glycolysis and
pentose phosphate metabolism, thereby suppressing the generation of
ATP, macromolecules, and NADPH required for cancer cell growth,
proliferation, and survival. Furthermore, our findings indicate
that ectopic overexpression of GLUT1, combined with galactose
supplementation to enhance PPP-mediated NADPH and biomass
generation, confers resistance to apigenin-mediated apoptosis in
several lung cancer cell lines. GLUT1 is upregulated in multiple
types of cancer, and is closely correlated with cancer grade,
radio-resistance, and chemo-resistance (10,37).
Indeed, several GLUT1 inhibitors, including WZB117 and fasentin,
have been developed as anticancer drugs (12,13).
Thus, apigenin treatment might provide a selective anticancer
strategy, acting through suppression of GLUT1-mediated cancer
growth and survival.
Growing cancer cells use large amounts of glucose
and glutamine to meet the bioenergetics and biosynthetic demands of
increased cell growth and survival (7,8). It
is becoming clear that altered metabolic pathways in cancer cells,
such as aerobic glycolysis, fatty acid synthesis, and glutamine
utilization are closely linked to therapeutic resistance in cancer
treatment (7,8,26).
Therefore, treatment of cancer using combinations of
chemotherapeutic drugs and selective cancer metabolism inhibitors
could represent a promising strategy to overcome chemoresistance.
Indeed, several reports have shown that inhibition of glycolysis
enhances the susceptibility of lung cancer, multiple myeloma, and
breast cancer to apoptosis during treatment with anticancer drugs,
such as cisplatin, doxorubicin, and trastuzumab (18). Previous reports have shown that GLS
inhibitors, such as CB-839 and compound 968, can suppress cancer
growth and survival in vitro and in vivo (20,38).
However, these studies did not address whether the combined
inhibition of glucose and glutamine utilization might represent a
promising strategy for cancer treatment. In this study, we
demonstrated that the combined suppression of glucose and glutamine
utilization, using a combination of apigenin and compound 968,
markedly decreased cell viability and increased apoptosis in lung
cancer cells. Thus, our findings suggest that the simultaneous
inhibition of multiple cancer metabolic pathways may provide a
promising strategy for cancer treatment.
Mechanistically, increased ROS is involved in
apigenin-induced apoptosis in prostate and colorectal cancer cells
(3,6). However, the molecular mechanism by
which apigenin increases intracellular ROS levels is not well
understood. Because suppression of glucose utilization is reported
to induce oxidative stress through inhibition of the PPP and NADPH
generation (27), we hypothesized
that apigenin might induce oxidative stress by the same mechanism.
Indeed, we found that apigenin significantly decreased glucose
utilization through suppression of GLUT1 expression, and
consequently decreased NADPH production, which led to increased ROS
levels. In addition, activation of NADPH production by galactose
supplementation, which can be entered into PPP, significantly
reversed the apigenin-induced ROS accumulation. These results
therefore suggest a possible mechanism by which apigenin induces
oxidative stress through inhibition of glucose utilization and
PPP-mediated NADPH production.
In conclusion, the major findings are that: i)
apigenin decreases glucose utilization, via glycolysis and the PPP,
through suppression of GLUT1 expression, thereby resulting in the
inhibition of cancer cell growth and survival; ii) activation of
glucose utilization and the PPP by GLUT1 overexpression and
galactose supplementation confer resistance to apigenin-induced
apoptosis by compensating NADPH production, which suppresses ROS
accumulation; iii) targeting glutamine utilization sensitizes lung
cancer cells to apigenin-induced apoptosis under conditions of
severe metabolic stress. These results may provide a promising
therapeutic strategy for cancer treatment.
Acknowledgements
This study was supported by the faculty research
fund of Konkuk University in 2013.
Abbreviations:
|
PPP
|
pentose phosphate pathway
|
|
GLUT1
|
glucose transporter 1
|
|
ROS
|
reactive oxygen species
|
|
GLS
|
glutaminase
|
|
NAC
|
N-acetyl-L-cysteine
|
|
DCF-DA
|
2′-7′-dichrolodihydrofluorescein
|
References
|
1
|
Li-Weber M: Targeting apoptosis pathways
in cancer by Chinese medicine. Cancer Lett. 332:304–312. 2013.
View Article : Google Scholar
|
|
2
|
Gupta SC, Kim JH, Prasad S and Aggarwal
BB: Regulation of survival, proliferation, invasion, angiogenesis,
and metastasis of tumor cells through modulation of inflammatory
pathways by nutraceuticals. Cancer Metastasis Rev. 29:405–434.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Banerjee K and Mandal M: Oxidative stress
triggered by naturally occurring flavone apigenin results in
senescence and chemotherapeutic effect in human colorectal cancer
cells. Redox Biol. 5:153–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan LP, Chou TH, Ding HY, Chen PR, Chiang
FY, Kuo PL and Liang CH: Apigenin induces apoptosis via tumor
necrosis factor receptor- and Bcl-2-mediated pathway and enhances
susceptibility of head and neck squamous cell carcinoma to
5-fluorouracil and cisplatin. Biochim Biophys Acta. 1820:1081–1091.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kachadourian R and Day BJ:
Flavonoid-induced glutathione depletion: Potential implications for
cancer treatment. Free Radic Biol Med. 41:65–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shukla S and Gupta S: Apigenin-induced
prostate cancer cell death is initiated by reactive oxygen species
and p53 activation. Free Radic Biol Med. 44:1833–1845. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vander Heiden MG: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vazquez F, Lim JH, Chim H, Bhalla K,
Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman
BM, et al: PGC1α expression defines a subset of human melanoma
tumors with increased mitochondrial capacity and resistance to
oxidative stress. Cancer Cell. 23:287–301. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Szablewski L: Expression of glucose
transporters in cancers. Biochim Biophys Acta. 1835:164–169.
2013.
|
|
11
|
Kim MS, Kwon JY, Kang NJ, Lee KW and Lee
HJ: Phloretin induces apoptosis in H-Ras MCF10A human breast tumor
cells through the activation of p53 via JNK and p38
mitogen-activated protein kinase signaling. Ann NY Acad Sci.
1171:479–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y, Cao Y, Zhang W, Bergmeier S, Qian
Y, Akbar H, Colvin R, Ding J, Tong L, Wu S, et al: A small-molecule
inhibitor of glucose transporter 1 downregulates glycolysis,
induces cell-cycle arrest, and inhibits cancer cell growth in vitro
and in vivo. Mol Cancer Ther. 11:1672–1682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wood TE, Dalili S, Simpson CD, Hurren R,
Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, et al: A
novel inhibitor of glucose uptake sensitizes cells to FAS-induced
cell death. Mol Cancer Ther. 7:3546–3555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hatanaka M: Transport of sugars in tumor
cell membranes. Biochim Biophys Acta. 355:77–104. 1974.PubMed/NCBI
|
|
15
|
Ulanovskaya OA, Cui J, Kron SJ and Kozmin
SA: A pairwise chemical genetic screen identifies new inhibitors of
glucose transport. Chem Biol. 18:222–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Budihardjo II, Walker DL, Svingen PA,
Buckwalter CA, Desnoyers S, Eckdahl S, Shah GM, Poirier GG, Reid
JM, Ames MM, et al: 6-Aminonicotinamide sensitizes human tumor cell
lines to cisplatin. Clin Cancer Res. 4:117–130. 1998.PubMed/NCBI
|
|
18
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seltzer MJ, Bennett BD, Joshi AD, Gao P,
Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS,
Rabinowitz JD, et al: Inhibition of glutaminase preferentially
slows growth of glioma cells with mutant IDH1. Cancer Res.
70:8981–8987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang JB, Erickson JW, Fuji R, Ramachandran
S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et
al: Targeting mitochondrial glutaminase activity inhibits oncogenic
transformation. Cancer Cell. 18:207–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim JH, Luo C, Vazquez F and Puigserver P:
Targeting mitochondrial oxidative metabolism in melanoma causes
metabolic compensation through glucose and glutamine utilization.
Cancer Res. 74:3535–3545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bao YY, Zhou SH, Fan J and Wang QY:
Anticancer mechanism of apigenin and the implications of GLUT-1
expression in head and neck cancers. Future Oncol. 9:1353–1364.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bao YY, Zhou SH, Lu ZJ, Fan J and Huang
YP: Inhibiting GLUT-1 expression and PI3K/Akt signaling using
apigenin improves the radiosensitivity of laryngeal carcinoma in
vivo. Oncol Rep. 34:1805–1814. 2015.PubMed/NCBI
|
|
24
|
Melstrom LG, Salabat MR, Ding XZ, Milam
BM, Strouch M, Pelling JC and Bentrem DJ: Apigenin inhibits the
GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt
pathway in human pancreatic cancer cells. Pancreas. 37:426–431.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu YY, Wu TT, Zhou SH, Bao YY, Wang QY,
Fan J and Huang YP: Apigenin suppresses GLUT-1 and p-AKT expression
to enhance the chemosensitivity to cisplatin of laryngeal carcinoma
Hep-2 cells: An in vitro study. Int J Clin Exp Pathol. 7:3938–3947.
2014.PubMed/NCBI
|
|
26
|
Hamanaka RB and Chandel NS: Targeting
glucose metabolism for cancer therapy. J Exp Med. 209:211–215.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lim JH: Zinc finger and BTB
domain-containing protein 3 is essential for the growth of cancer
cells. BMB Rep. 47:405–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Le A, Lane AN, Hamaker M, Bose S, Gouw A,
Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al:
Glucose-independent glutamine metabolism via TCA cycling for
proliferation and survival in B cells. Cell Metab. 15:110–121.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wise DR and Thompson CB: Glutamine
addiction: A new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Agullo G, Gamet-Payrastre L, Manenti S,
Viala C, Rémésy C, Chap H and Payrastre B: Relationship between
flavonoid structure and inhibition of phosphatidylinositol
3-kinase: A comparison with tyrosine kinase and protein kinase C
inhibition. Biochem Pharmacol. 53:1649–1657. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang J, Xia C, Cao Z, Zheng JZ, Reed E and
Jiang BH: Apigenin inhibits VEGF and HIF-1 expression via
PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 19:342–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang YT, Kuo ML, Liu JY, Huang SY and Lin
JK: Inhibitions of protein kinase C and proto-oncogene expressions
in NIH 3T3 cells by apigenin. Eur J Cancer. 32A:146–151. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Llorens F, Miró FA, Casañas A, Roher N,
Garcia L, Plana M, Gómez N and Itarte E: Unbalanced activation of
ERK1/2 and MEK1/2 in apigenin-induced HeLa cell death. Exp Cell
Res. 299:15–26. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yin F, Giuliano AE, Law RE and Van Herle
AJ: Apigenin inhibits growth and induces G2/M arrest by modulating
cyclin-CDK regulators and ERK MAP kinase activation in breast
carcinoma cells. Anticancer Res. 21(1A): 413–420. 2001.PubMed/NCBI
|
|
37
|
Adekola K, Rosen ST and Shanmugam M:
Glucose transporters in cancer metabolism. Curr Opin Oncol.
24:650–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gross MI, Demo SD, Dennison JB, Chen L,
Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et
al: Antitumor activity of the glutaminase inhibitor CB-839 in
triple-negative breast cancer. Mol Cancer Ther. 13:890–901. 2014.
View Article : Google Scholar : PubMed/NCBI
|