1. Introduction
Cancer is defined by the American Cancer Society as
a group of diseases characterized by the uncontrolled growth and
spread of abnormal cells. The World Health Organization (WHO)
states that one defining feature of cancer is the rapid creation of
abnormal cells that grow beyond their usual boundaries, and which
can then invade adjoining parts of the body and spread to other
organs. This growth is caused by either external factors (tobacco,
infectious organisms and an unhealthy diet), or by internal factors
(inherited genetic mutations, hormones, and immune conditions)
(https://www.cancer.org).
Cancer can have severe health consequences, and is a
leading cause of mortality worlwide; there are 8.2 million
cancer-related deaths, which corresponds to 13% of all deaths
worldwide. Data from the Cancer Research UK indicated that in 2012,
approximately 14.1 million new cases of cancer were diagnosed in
worldwide. Among these cases, 7.4 million (53%) were diagnosed in
males and 6.7 million (47%) in females, with a male-to-female ratio
of 10:9. The world age-standardized incidence rate points out that
there are 205 new cancer cases for every 100,000 males worldwide,
and 165 for every 100,000 females (1).
According to WHO, lung, prostate, colorectal,
stomach and liver cancer are the most common types of cancer in
males, while breast, colorectal, lung, uterine cervix and stomach
cancer are the most common among females. Statistics also indicate
that there is expected to be a 70% increase in new cancer cases
over the next two decades. The American Cancer Society released the
analysis for 2015, projecting that there will be an estimated
1,658,370 new cancer cases diagnosed and 589,430 cancer-related
deaths in the US (2).
Presently, there are over 100 types of cancer, which
require a great effort for diagnosis and treatment; due to this
high incidence and high risk for public health, intense research
designed into this topic is being carried out worldwide.
Over the past decades, a growing body of evidence
has indicated that cancer patients can be cured by novel molecular
target therapies due to the molecular characterization of the tumor
from each patient. The phosphatidylinositol 3-kinase (PI3K)/Akt
pathway is aberrantly activated in several types of cancers and
targeting this pathway with drug inhibitors may result in more
effective anticancer treatment for both solid and hematologic
tumors (3). In this review, recent
data regarding the regulation of the PI3K/Akt signal transduction
pathway and its involvement in the development and progression
cancer are discussed. Furthermore, the most relevant studies
focusing on the specific action of new molecular targeted agents
are discussed.
2. Akt structure and function
The serine/threonine kinase Akt, also known as
protein kinase B or PKB, with three isoforms (Akt1, Akt2 and Akt3),
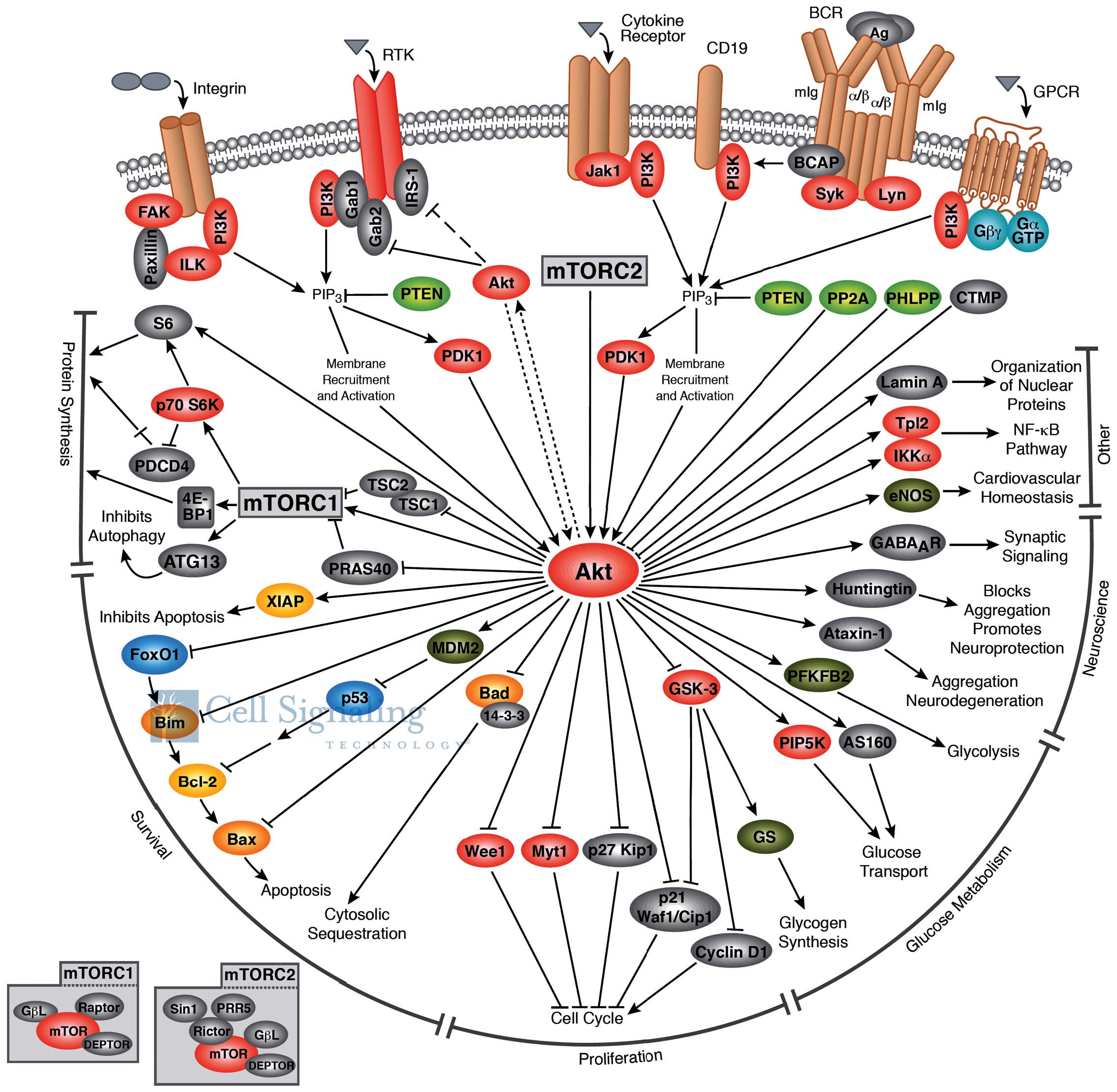
plays a critical role in regulating diverse cellular functions
(Fig. 1) including cell
size/growth, proliferation, survival, glucose metabolism, genome
stability, transcription and protein synthesis, and
neovascularization (4). One of the
major functions of Akt/PKB is to promote growth factor-mediated
cell survival, to promote cell proliferation and to inhibit
apoptosis through the inactivation of pro-apoptotic proteins, such
as Bad (Bcl-2 antagonist of cell death) and mouse double minute 2
homolog (MDM2; which causes the degradation of p53) (5).
Studies have proven that the Akt signaling cascade
is frequently impaired in many types of cancer and, in some cases,
that it is associated with tumor aggressiveness. These are the
reasons for considering this signaling pathway as a strong target
candidate for cancer therapy or even cancer prevention (6,7).
The Akt structure consists of a three domains: i) an
amino terminal pleckstrin homology (PH) domain, consisting of 100
amino acids, sharing similarity to those found in other signaling
molecules binding 3-phosphoinositide; this domain interacts with
membrane lipid products, such as
phosphatidylinositol-3,4,5-triphosphate (PIP3) and
phosphatidylinositol 4,5-bisphosphate (PIP2); ii) a central kinase
domain, highly similar to other cAMP-dependent protein kinase C
(AGC kinases), containing a regulatory threonine residue (Thr308)
which phosphorylation activates Akt; iii) a carboxyl-terminal
regulatory domain containing the hydrophobic region consisting of
40 amino acids, including the serine regulatory residue (Ser473)
(8).
Akt isoforms share a high degree of sequence
homology in their catalytic domains, but diverge in the regulatory
domain and the PH domain. Akt1 and Akt2 are ubiquitously expressed,
whereas Akt3 is found predominantly in the brain, heart and kidneys
(9). The catalytic domain of Akt
displays a high degree of similarity to those found in
cAMP-dependent protein kinase or protein kinase A (PKA) and protein
kinase C (PKC), hence the other name of Akt, protein kinase B
(PKB). Structurally, and functionally, Akt belongs to the AGC
kinase family, a group named after its major representatives, the
PKA, the cGMP-dependent protein kinase or protein kinase G (PKG)
and PKC (10).
Akt, together with PI3K, are the key elements of the
Akt signaling cascade, also known as PI3K/Akt, a signal
transduction pathway that promotes survival and growth in response
to extracellular signals (5). This
signaling cascade can be activated by receptor tyrosine kinases,
integrins, B and T cell receptors, cytokine receptors,
G-protein-coupled receptors and other stimuli that induce
production of PIP3 (11,12). PI3K is overexpressed in ovarian and
cervical cancer and has mutations associated with breast cancer,
glioblastoma and gastric cancer (13,14).
The activation of Akt firstly involves a PH
domain-dependent membrane translocation step, followed by the
phosphorylation of the two key regulatory sites, Thr308 and Ser473
(15). Various signaling events
activate PI3K to phosphorilate its membrane substrate, PIP2,
generating PIP3 (16). PIP3
recruits Akt to the plasma membrane by interacting with the PH
domain and modifies its conformation to allow subsequent
phosphorylation by the phosphoinositide-dependent kinase-1 (PDK1)
at the Thr308 site in the regulatory domain (17). This signaling event primes Akt for
phosphorylation at Ser473 by mechanistic target of rapamycin
(mTOR)C2 (16). Members of the
PI3K-related kinase family, including DNA-PK, can also
phosphorylate Akt at Ser473 (18).
In addition, there are some binding proteins which regulate the
activity of Akt [actin, extracellular signal-regulated protein
kinase (Erk)1/2, heat shock protein (Hsp)90, Hsp27 and Posh]. For
example, Hsp90 uses repeated cycles of client protein binding, the
hydrolysis of adenosine triphosphate (ATP) and interaction with its
co-chaperones, such as Hsp70, Cdc37, HOP, p23 and activator of heat
shock 90 kDa protein ATPase homolog 1 (Aha1) to control the
stability and activity of hundreds of client proteins involved in
critical signaling pathways necessary for cellular proliferation,
cell cycle progression, and apoptosis, including Akt (19).
Akt is dephosphorylated by protein phosphatase 2A
(PP2A) and the PH domain leucine-rich-repeat-containing protein
phosphatase 1/2 (PHLPP2). In addition, the tumor suppressor,
phosphatase and tensin homolog (PTEN), inhibits Akt activity by
dephosphorylating PIP3 (20).
Activated Akt modulates the function of numerous
substrates involved in the regulation of cell survival, cell cycle
progression and cellular growth (21). Akt modulates cellular processes by
phosphorylating different substrates, such as proline-rich Akt
substrate of 40 kDa (PRAS40), actin-associated protein paladin, CDK
inhibitors p21 and p27, palladin and vimentin, IKKα and Tpl2
(9). Akt regulates cell growth
(Fig. 1) through its effects on
the TSC1/TSC2 complex and mTORC signaling and acts as a major
mediator of cell survival through the direct inhibition of
pro-apoptotic proteins, such as Bad or through the inhibition of
pro-apoptotic signals generated by transcription factors, such as
Forkhead box protein O1 (FoxO) (22).
mTOR is a downstream member of the PI3K/Akt and
adenosine monophosphate-activated protein kinase pathways, and is a
key regulator of cell growth and metabolism. mTOR (Fig. 1) is a component of two similar
complexes: mTORC1, which promotes mRNA translation and protein
synthesis by the phosphorylation of ribosomal protein S6 kinase
(S6K1) and eIF4E binding protein 1 (4E-BP1) and mTORC2, which
organizes the cellular actin cytoskeleton and regulates Akt
phosphorylation (22).
As Akt isoforms share a high degree of structural
similarity, it has long been assumed that their functions largely
overlap. However, Akt1 has a cardio-protective role by supporting
heart's physiological growth and function (23). Strong evidence indicates a
significant association between schizophrenia and a decrease in
Akt1 levels (24). As previously
demonstrated, hyperglycemia and reduced glucose transport in muscle
are present in knockout Akt2 mice, but not following the depletion
of Akt1 or Akt3 (25). A recent
study indicated that both Akt1 and Akt2 contribute to
insulin-dependent glucose transporter type 4 (GLUT4) translocation
to the membrane (26). Akt3 does
not appear to significantly influence normal metabolism, but it is
critical for brain development; several Akt3 mutations have been
reported in neurological disorders (27).
3. Akt inhibitors as anticancer agents
The Akt pathway is one of the most frequently
deregulated signaling pathways in human cancers with many of its
components described as altered. The activation of the Akt pathway
plays an essential role in cell survival, proliferation, migration
and differentiation, contributing to tumorigenesis and tumor
metastasis. The overexpression and activation of Akt are often
associated with resistance to chemotherapy or radiotherapy
(28). Our preliminary results,
which are still unpublished, suggest that Akt activation is
observed in melanoma samples from patients with occupational sun
exposure. Such an Akt activation has also been identified in
melanoma samples, previously screened for B-RAFV600E
mutations (29), harboring both
B-RAFV600E and PI3KH1047R. Intriguingly, both
mutations were detected among indoor workers with intermittent sun
exposure, suggesting that Akt activation may be linked with a
previous damage to DNA by ultraviolet (UV) light.
Akt is also overexpressed or activated in a variety
of human cancers, including lung, breast, ovarian, gastric and
pancreatic carcinomas (30). PTEN
activity can be impaired by mutations, deletions or promoter
methylation in many primary and metastatic human cancers, and
activating mutations of PI3K have been observed in various human
tumors (31). It has been
demonstrated that a low PTEN expression correlates with poor
responses to trastuzumab-based therapy (33) and it has been hypothesized that Akt
inhibitors may be useful in these cases. Various substances that
can also increase PTEN levels are under investigation as anticancer
therapies, such as the proteasome inhibitor, bortezomib (34), and the natural lignans,
deoxypodophyllotoxin (35) and
matairesinol (36).
An oncogenetic activating mutation (E17K) in the PH
domain of Akt1 has been detected in some types of solid tumors.
This mutation has been reported in breast (5.9%), colorectal
(1.6%), lung (0.6%) and ovarian cancers (0.8%), as well as in
melanoma (0.5%) (37). Akt1 is
frequently elevated in breast and prostate cancers. The
amplification and overexpression of Akt2 has been shown to
correlate with the aggressiveness of cancer and poor survival
rates, and is frequently detected in prostate, ovarian, breast,
pancreatic and colorectal cancers (38–40).
In previous studies, the immunohistochemical staining of clinical
samples revealed that Akt3 protein expression was upregulated in
androgen-independent prostate cancer cell lines, estrogen
receptor-deficient breast cancer cells (41) and in primary ovarian cancers
(42).
Akt is considered as an attractive target for cancer
therapy and multiple attempts to identify specific inhibitors with
acceptable pharmaceutical properties have been pursued. Despite
significant progress being made, selectivity is a key issue for
many ATP-competitive Akt inhibitors, particularly towards the AGC
kinase family. The development of Akt inhibitors has been also
hampered by the existence of three isozymes, which differ in
function, tissue distribution and affinity for ligands (43). The development of Akt-specific and
isoform-selective inhibitors using the catalytic domain of the
kinase has been predicted to be difficult due to high sequence
homology, determining alternative and novel approaches to target
Akt and to identify allosteric inhibitors (44).
The major Akt inhibitors classified by the
inhibition mechanisms and by their chemical scaffold are presented
in Table I.
 | Table IAkt-inhibiting drugs listed into
major classes. |
Table I
Akt-inhibiting drugs listed into
major classes.
| Class | Description |
|---|
| ATP-competitive
inhibitors | Orthosteric
inhibitors targeting the ATP-binding pocket of the protein kinase B
(Akt) |
|
Isoquinoline-5-sulfonamides | H-8, H-89,
NL-71-101 |
| Azepane
derivatives | A series structures
derived from (−)-balanol |
| Aminofurazans | GSK690693 |
| Heterocyclic
rings | 7-azaindole,
6-phenylpurine derivatives, pyrrolo[2,3-d]pyrimidine derivatives,
CCT128930, 3-aminopyrrolidine, anilinotriazole derivatives,
spiroindoline derivatives, AZD5363, ipatasertib (GDC-0068, RG7440),
A-674563, A-443654 |
| Phenylpyrazole
derivatives | AT7867,
AT13148 |
|
Thiophenecarboxamide derivatives | Afuresertib
(GSK2110183), 2-pyrimidyl-5-amidothiophene derivative (DC120),
uprosertib (GSK2141795) |
| Allosteric
inhibitors | Superior to
orthosteric inhibitors providing greater specificity, reduced
side-effects and less toxicity |
|
2,3-diphenylquinoxaline analogues |
2,3-diphenylquinoxaline derivatives,
triazolo[3,4-f][1,6]naphthyridin-3(2H)-one derivative
(MK-2206) |
|
Alkylphospholipids | Edelfosine
(1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine,
ET-18-OCH3) ilmofosine (BM 41.440), miltefosine
(hexadecylphosphocholine, HePC), perifosine (D-21266),
erucylphosphocholine (ErPC), erufosine (ErPC3,
erucylphosphohomocholine |
| Indole-3-carbinol
analogues | Indole-3-carbinol,
3-chloroacetylindole, diindolylmethane, diethyl
6-methoxy-5,7-dihydroindolo
[2,3-b]carbazole-2,10-dicarboxylate (SR13668), OSU-A9 |
| Sulfonamide
derivatives | PH-316,
PHT-427 |
| Thiourea
derivatives | PIT-1, PIT-2,
DM-PIT-1,
N-[(1-methyl-1H-pyrazol-4-yl)carbonyl]-N′-(3-bromophenyl)-thiourea |
| Purine
derivatives | Triciribine (TCN,
NSC 154020), triciribine mono-phosphate active analogue (TCN-P),
4-amino-pyrido[2,3-d]pyrimidine derivative API-1,
3-phenyl-3H-imidazo[4,5-b]pyridine derivatives, ARQ
092 |
| Other structures,
derivatives | BAY 1125976,
3-methyl-xanthine, quinoline-4-carboxamide and
2-[4-(cyclohexa-1,3-dien-1-yl)-1H-pyrazol-3-yl]phenol,
3-oxo-tirucallic acid, 3α- and 3β-acetoxy-tirucallic acids,
acetoxy-tirucallic acid |
| Irreversible
inhibitors | Natural products,
antibiotics
Lactoquinomycin, Frenolicin B, kalafungin, medermycin,
Boc-Phe-vinyl ketone,
4-hydroxynonenal (4-HNE), 1,6-naphthyridinone derivatives,
imidazo-1,2-pyridine derivatives |
4. ATP-competitive inhibitors
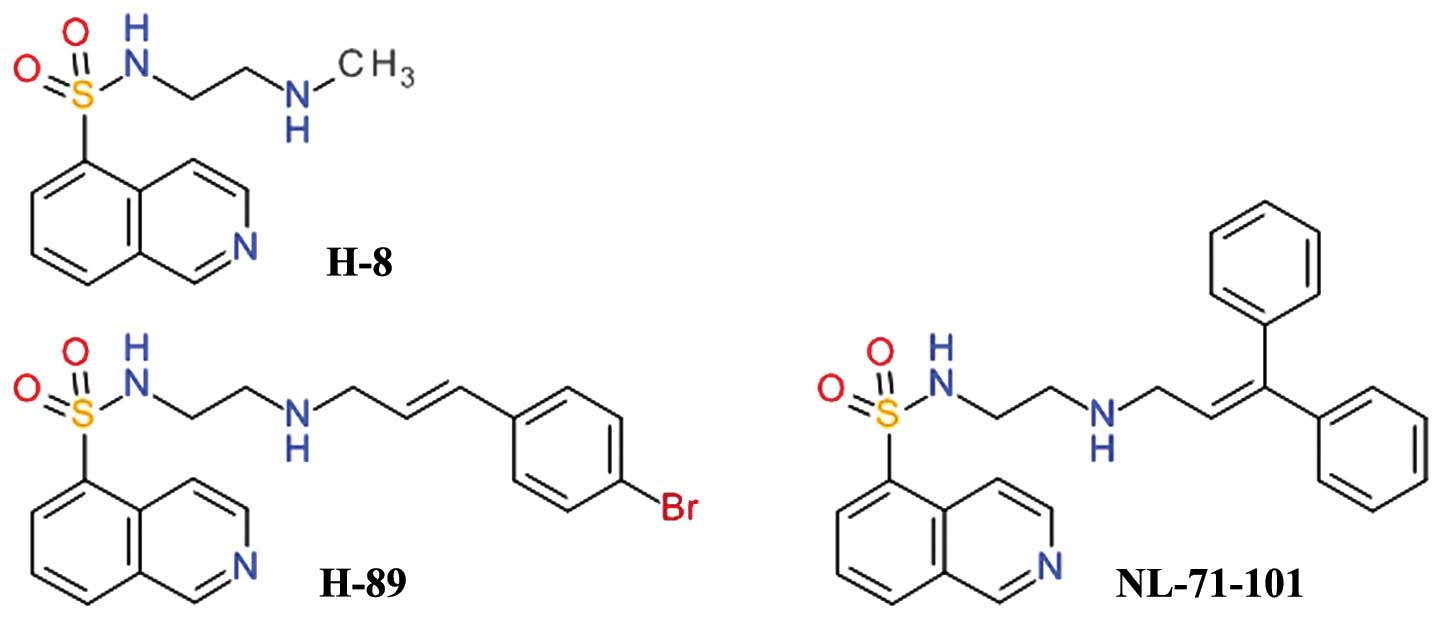
Isoquinoline-5-sulfonamides
One of the first relatively selective inhibitors of
Akt was NL-71-101, obtained after the screening of a combinatorial
library designed using the structure of the
isoquinoline-5-sulfonamide derivative, H-89, as a starting point
(Fig. 2). H-89 is a potent
inhibitor of PKA and it was observed to inhibit the growth of the
human colon cancer cell line, Caco-2, in a dose-dependent manner,
while the congener derivative, H-8, did not. H-89 proved to be an
inhibitor of Akt and opened a new direction for development of Akt
inhibitors (45). H-89 has been
shown to inhibit PKA 70 - fold more potently than Akt, but
NL-71-101 is relatively selective towards Akt, inhibiting it
2–3-fold more potently than PKA (46). This compound induces the apoptosis
of ovarian carcinoma cells at high concentrations (43).
Azepane derivatives
A similar strategy was used to improve the
inhibition of Akt over PKA, starting from the natural fungal
metabolite, (−)-balanol. The ester moiety of the balanol is
responsible for its low stability and was replaced with an amide
group rendering a new series of azepane inhibitors of Akt (47). Most programs to develop
ATP-competitive inhibitors of Akt are driven by achieving
selectivity towards PKA because of its role over key cell functions
(48).
Aminofurazans
GSK690693 is an aminofurazan derivative,
ATP-competitive inhibitor with IC50 values of 2, 13 and
9 nM against Akt 1, 2 and 3, respectively (49). In preclinical studies, GSK690693
was shown to inhibit the proliferation of various hematologic
neoplasia, with acute lymphoblastic leukemia cell lines being the
most sensitive (50), but exerted
limited effects on xenograft models (51). The clinical development of this
compound was terminated due to the associated side-effect of
transient hyperglycemia (52).
Compounds with heterocyclic 6-5 fused
rings
Virtual, biochemical and crystallographic screens
identified 7-azaindole as an important small fragment for the
design of Akt inhibitors. The optimization of the scaffold advanced
to the development of 6-phenylpurine (53) and afterwards to
pyrrolo[2,3-d]pyrimidine derivatives. The coupling of the
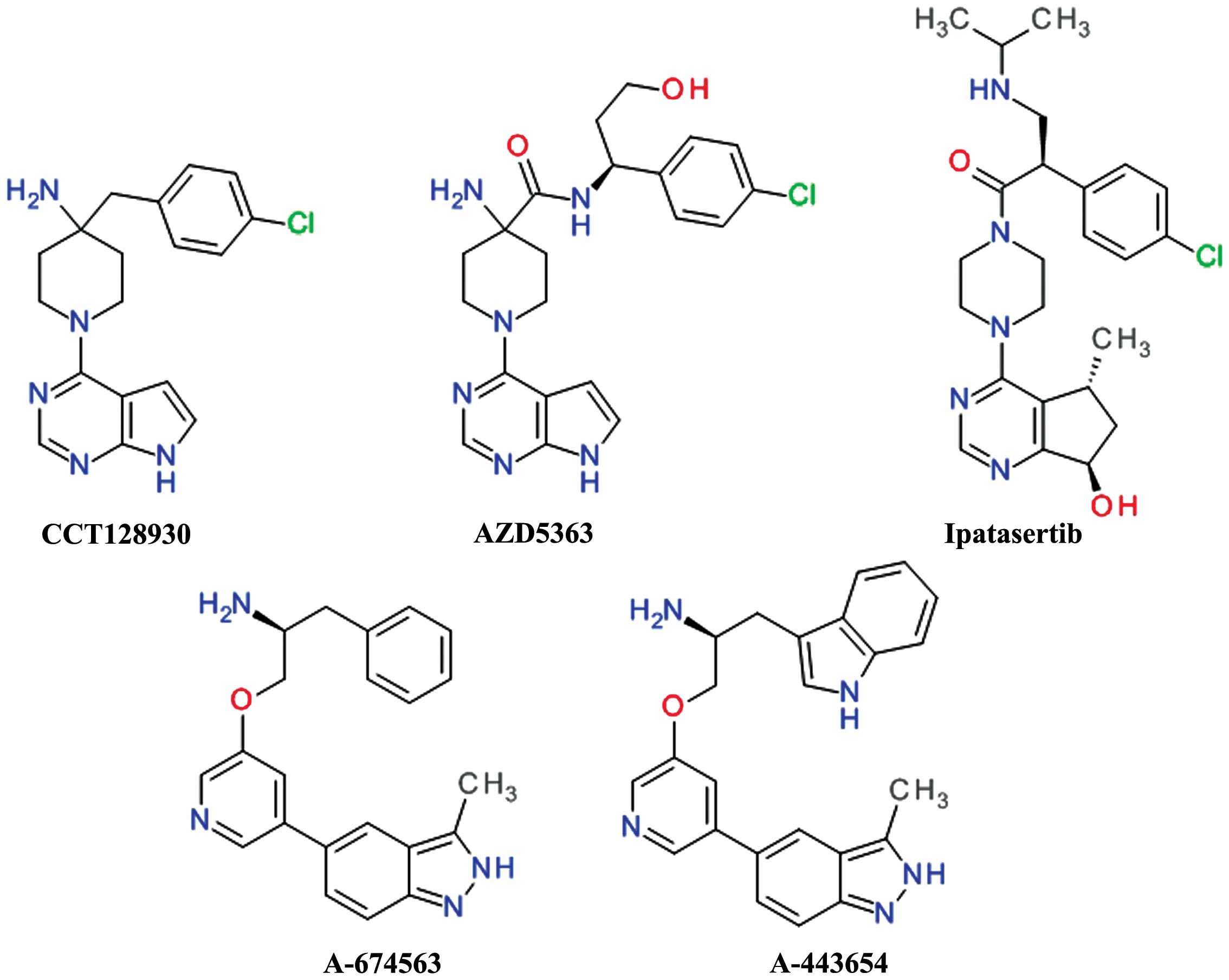
4-aminopiperidine and pyrrolopyrimidine moieties afforded CCT128930
and provided selectivity over PKA (54). CCT128930 (Fig. 3) has been shown to inhibit the
phosphorylation of a range of Akt substrates and to induce marked
antitumor responses in various cancer cell lines (55). The pyrrolopyrimidine scaffold was
used extensively in the development of various Akt inhibitors. The
replacement of the piperidine ring in the structure of CCT128930
has led to a series of 3-aminopyrrolidines with a very good
selectivity over PKA (56). A high
throughput in vitro kinase assay of pyrrolopyrimidines has
led to the development of a potent Akt1 inhibitor with an
anilinotriazole structure. Further development replaced the
anilinotriazole moiety with an imidazopiperidine which led to the
discovery of the spiroindolines scaffold as linker to the
pyrrolo[2,3-d]pyrimidine (57).
In order to improve the rapid in vivo
metabolism and low oral bioavailability of CCT128930, the
4-amino-4-benzylpiperidine moiety was modified to
4-amino-piperidine-4-carboxamide, leading to the development of
AZD5363 (58).
AZD5363 (Fig. 3)
has been shown to inhibit all Akt isoforms with a potency of ≤10
nmol/l and has shown reduced hERG affinity, and a higher
selectivity against the closely related Rho-associated protein
kinase (ROCK) in addition to good pharmacokinetics properties
(59). Treatment with AZD5363 has
been shown to inhibit the proliferation of 41 of 182 solid and
hematological tumor cell lines with a potency of ≤3 μmol/l and the
highest frequency of sensitivity was observed in breast cancer
cells. AZD5363 significantly enhanced the antitumor activity of
docetaxel, lapatinib and trastuzumab in breast cancer xenografts.
The activity of AZD5363 in human epidermal growth factor receptor 2
(HER2)-amplified breast cancer cells was enhanced by the addition
of a pan-erbB [epidermal growth factor receptor (EGFR)] tyrosine
kinase (60). Several clinical
assays of phase I and II are undergoing to assess the AZD5363 in
breast, gastric and prostate cancers (61).
Ipatasertib (GDC-0068, RG7440) was the result of the
discovery and optimization of a series of
6,7-dihydro-5H-cyclopenta[d]pyrimidine compounds. It is
orally bioavailable and has demonstrated potent inhibition of all
three Akt isoforms and a poor inhibition of other members of the
PKA family (62). Treatmetn with
ipatasertib has been shown to inhibit Akt signaling in both human
cancer cell lines and in tumor xenograft models, resulting in the
blockade of cell cycle progression the decreased viability of
cancer cell lines (63).
Ipatasertib is currently under evaluation in several clinical phase
I and II trials (NCT02430363 and NCT02301988). In a phase Ib
clinical study, the combination of ipatasertib with paclitaxel was
well-tolerated and a phase II study designed to estimate the
efficacy of this combination in metastatic triple-negative breast
cancer patients is in progress (64).
A-674563 and A-443654 (Fig. 3) were the result of a
high-throughput screening hit which weakly inhibited Akt. The
potency was improved by the addition of an indole ring to the
aliphatic side chain and by constraining rotatable bonds between
the central and distal pyridine rings by forming an isoquinoline
ring. The isoquinoline moiety was transformed in an indazole ring
to afford a second hydrogen interaction to the hinge region,
resulting in A-443654. The replacement the indole with a phenyl
moiety resulted in compound A-674563 and provided oral
bioavailability (65). These
compounds are potent, ATP-competitive and reversible inhibitors of
Akt. A-443654 has equal potency against Akt isoforms and is 40-fold
more selective for Akt over PKA, while A-674563 is less selective,
particularly against the cyclin-dependent kinases (66). Both compounds behave similarly
in vivo antitumor activity assay, with A-443654 being more
potent and more selective than A-674563. The both derivatives
increased the efficacy of paclitaxel in a prostate carcinoma cell
xenograft model (65).
Several other nitrogen compounds containing 6-5
fused rings have proven to be good templates for the development of
potent Akt inhibitors. Azaindazole and 4,7-diazaindazole were
incorporated in the structure of tetrasubstituted aminopyridines
providing potent and selective Akt inhibitors (67). A series of
1H-indazole-4,7-diones have been shown to exert dual
inhibitory effects on both the activity and phosphorylation of Akt1
in the PC-3 tumor cell line (68).
Dihydrothieno- and dihydrofuro- pyrimidine scaffolds generated
potent pan Akt inhibitors (69).
GSK690693, described above and classified as an aminofurazane
derivative, can also be included in the 6-5 fused rings group based
on its imidazo[4,5-c]pyridine core.
Phenylpyrazole derivatives
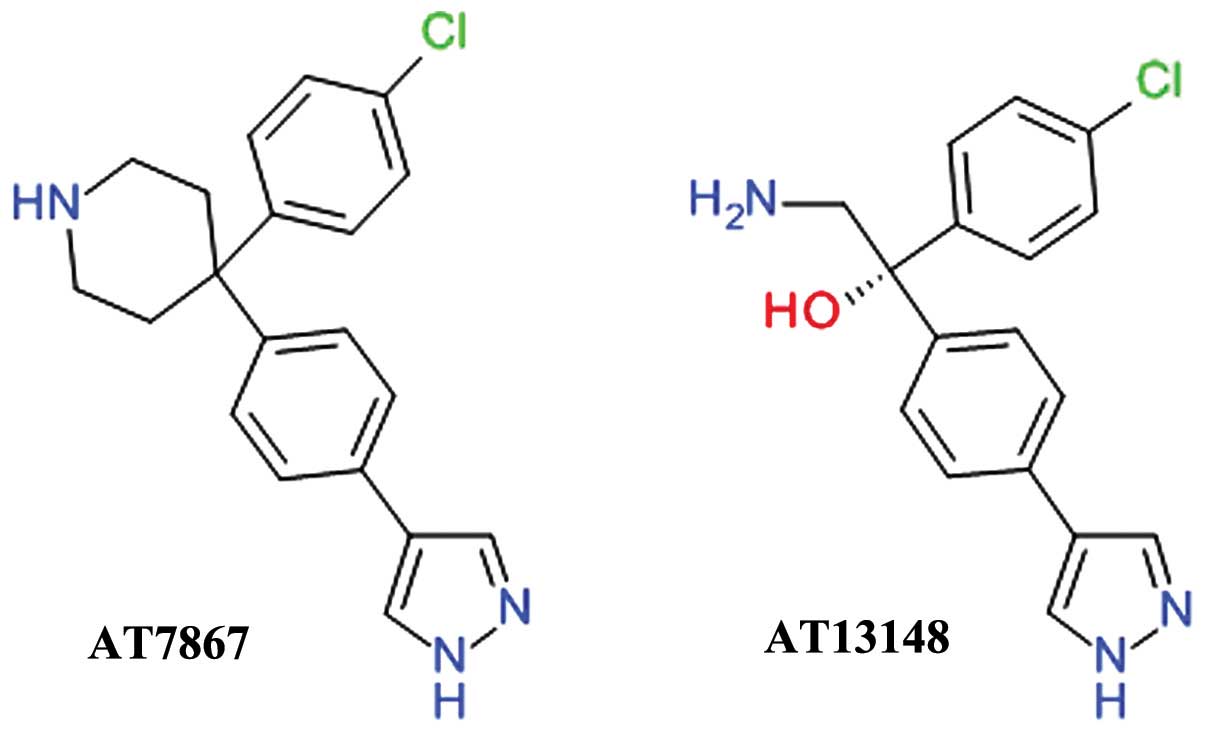
AT7867 was developed using fragment- and
structure-based drug design starting from the discovery of
4-phenylpyrazole as week Akt inhibitor. The inhibition of Akt2 by
AT7867 was shown to be ATP-competitive and the binding at the ATP
site was confirmed by determining the three-dimensional structure
of the ligand-enzyme complex using X-ray crystallography (70). AT7867 (Fig. 4) is an oral potent inhibitor of all
Akt isoforms and of the downstream p70 S6 kinase (p70 S6K) and also
of PKA. It suppresses cell proliferation, and induces the apoptosis
of a range of human cancer cell lines, but is still in preclinical
studies (71).
AT13148 (Fig. 4)
was identified using high-throughput X-ray crystallography and
fragment-based lead discovery techniques starting from a
4-phenylpyrazole scaffold, such as AT7867. Both compounds share a
high degree of structural similarity and, accordingly, comparable
inhibitions profiles. AT13148 is an ATP-competitive multi-AGC
kinase inhibitor blocking Akt, p70 S6K, PKA, ROCK, and serum and
glucocorticoid-inducible kinase (SGK). It has a marked apoptotic
rather than cytostatic profile, emphasizing the functional
differences between its properties as a multi-kinase inhibitor in
contrast with the selective Akt inhibitors (72).
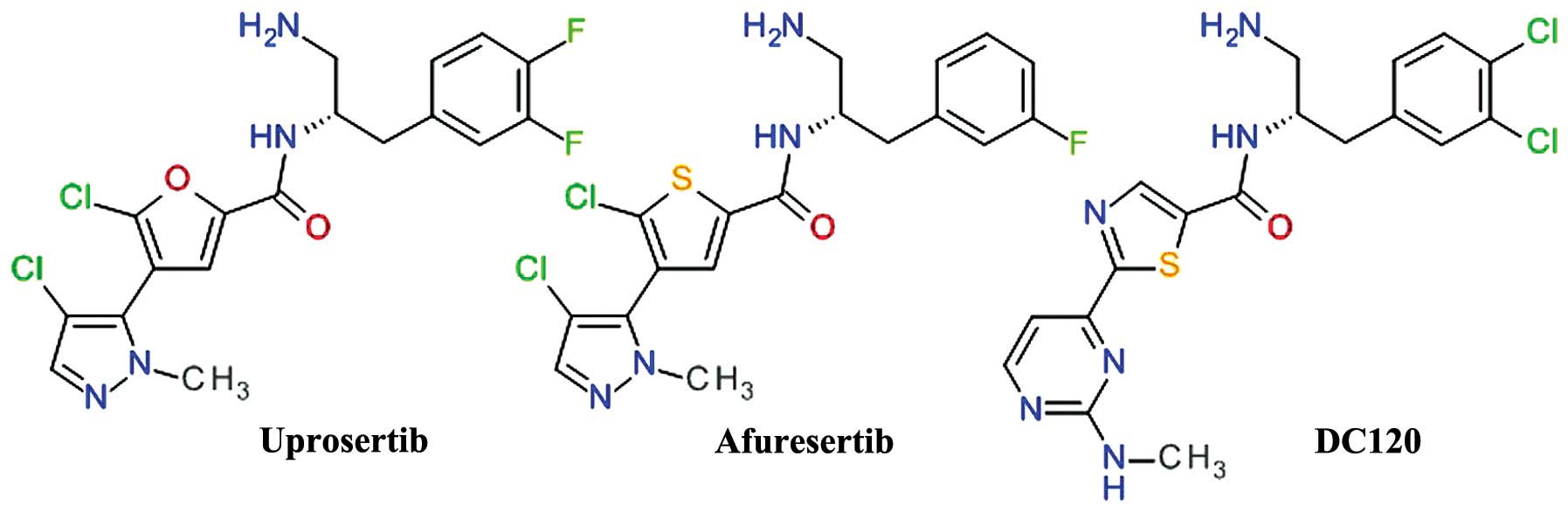
Thiophenecarboxamides and
derivatives
A derivate of 2-pyrimidyl-5-amidothiophene was
identified as an ATP-competitive inhibitor of Akt3 using high
throughput screening. The lead optimization based on
structure-activity associations demonstrated the importance of the
amide bond and the optimal length of two carbon atoms between the
phenyl ring and the amide nitrogen (73). The replacement of the pyrimidine
moiety proved to be useful, especially with a pyrazole ring,
resulting in the development of afuresertib (GSK2110183) (74). In the series of
2-pyrimidyl-5-amidothiophene derivatives, the transformation of the
thiophene in thiazole afforded a new lead compound DC120 (75). For both related compounds,
afuresertib and DC120 (Fig. 5),
the stereochemistry of the isomers is very important for the enzyme
inhibition.
DC120 has been shown to inhibit the proliferation of
CNE2 and MDA-MB-453 cells via the induction of apoptosis mediated
by cleaved caspase-3 and reduced the phosphorylation levels of
forkhead transcription factor (FKHR), glycogen synthase kinase 3β
(GSK-3β) and mTOR in a dose- and time-dependent manner. In
xenograft models, DC120 inhibited CNE2 tumor growth (76).
Afuresertib (GSK2110183) is a highly potent
inhibitor of Akt, with subnanomolar potency against Akt1 and low
nanomolar potency against Akt2 and Akt3. Proliferation assays of
human tumor cell lines demonstrated that the hematological cell
lines were the most sensitive, mostly acute lymphoblastic leukemia,
non-Hodgkin's lymphoma and chronic lymphocytic leukemia (74). In a clinical study, afuresertib
(PKB112835) was shown to be safe and well-tolerated. It produced a
relatively low incidence and magnitude of hyperglycemia, due to the
improved kinase selectivity versus other PKC isoforms. Afuresertib
proved its clinical efficacy in a number of hematological
malignancies, particularly in multiple myeloma (77). A phase I/II study on afuresertib in
combination with bortezomib and dexamethasone in patients with
relapsed or refractory myeloma showed an overall response rate of
41% (78).
Uprosertib (GSK2141795) is a structurally close
analog of afuresertib, the main difference being the replacement of
the thiophene core with its bioisostere furan ring (Fig. 5). The inhibition of Akt and
anti-proliferative effect of uprosertib is similar to that of
afuresertib, with uprosertib being more potent and having greater
off-target kinase inhibition. It is was tested clinically alone or
in combination with trametinib (MEK inhibitor) in various types of
cancer (74).
5. Allosteric inhibitors
ATP-competitive inhibitors are non-selective against
Akt isozymes, and poorly selective against closely related kinases,
as the catalytic domain is highly similar. Efforts to identify
Akt-specific and isoform-selective inhibitors have resulted in the
discovery of allosteric inhibitors (79). Allosteric modulators offer distinct
advantages compared to orthosteric ligands that target to active
sites, such as greater specificity, reduced side-effects and lower
toxicity (80).
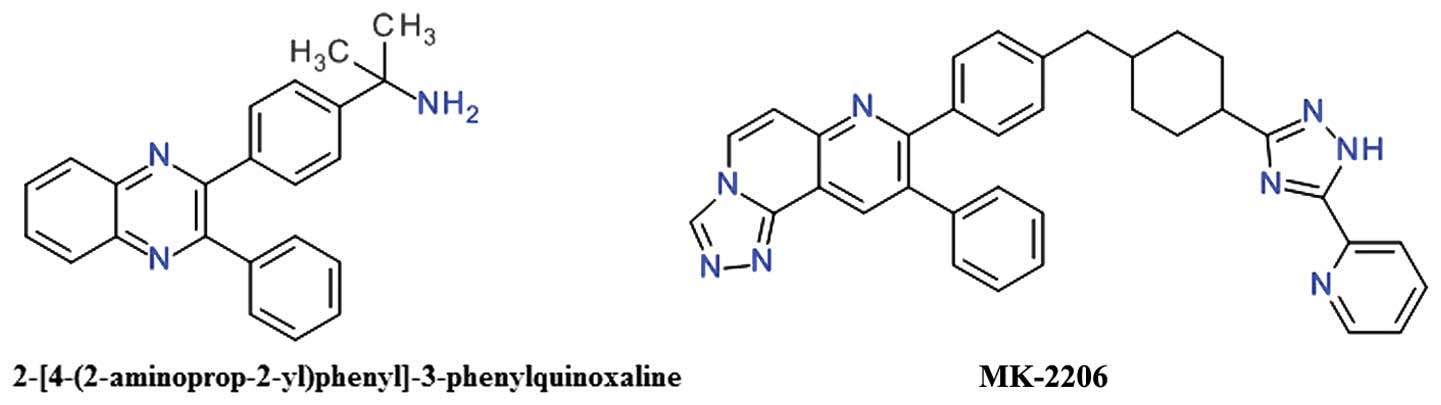
2,3-Diphenylquinoxaline and analogs
One of the first allosteric inhibitors of Akt was a
2,3-diphenylquinoxaline derivative that demonstrated potent
inhibitory effects against Akt1, but no effect on the mutated Akt
enzyme depleted of the PH domain (81). The lead compound (Fig. 6) resulted from a high throughput
screening focused to identify compounds capable of inhibiting all
three Akt isozymes, and was found to be selective over PKA, PKC and
SGK (81).
The 5,6-diphenyl-pyrazin-2(1H)-one scaffold
emerged as an analog of the 2,3-diphenylquinoxaline template and
was used to develop potent selective inhibitors for Akt1 and Akt2
(81). Imidazoquinoxaline also
proved to be a good template for the design of allosteric Akt
inhibitors (79).
The transformation of the core 1,4-quinoxaline with
a naphthyridine scaffold improved the compounds basicity and
therefore increased polarity and the solubility (82). The introduction of the
naphthyridine ring in a tricyclic structure significantly improved
Akt2 inhibition and cellular activities (83). MK-2206 is a
triazolo[3,4-f][1,6]naphthyridin-3(2H)-one derivative
(Fig. 6), is an orally active,
highly potent and selective inhibitor of all three Akt isoforms,
resulting in decreased p-Akt Thr308 and p-Akt Ser473 levels, and
consequently, in the reduction of the phosphorylation of the
downstream targets, GSK-3β, PRAS40, FoxO1/FoxO3a, and Bad (84). It binds in a pocket formed at the
interface of the catalytically active kinase domain and the
regulatory PH domain locking the kinase in a closed conformation
and preventing the adaptation of the active state (85).
MK-2206 induces G1-phase cell-cycle arrest and
increased apoptosis, accompanied by pro-caspase cleavage in a
concentration-dependent manner (86). Oxidation mediated by CYP3A4 is the
primary elimination pathway (87).
In preclinical experiments, MK-2206 demonstrated significant
synergy when combined with doxorubicin, camptothecin (topoisomerase
inhibitors), gemcitabine and 5-fluorouracil (antimetabolites),
carboplatin (DNA cross-linker), erlotinib (EGFR) and lapatinib
(dual EGFR/HER2 inhibitor) in lung NCI-H460 or ovarian A2780 cancer
cell lines (84). MK-2206 has been
extensively investigated, alone or in combination, in a
considerable number of phase I or II clinical trials. Several phase
I studies are evaluating MK-2206 alone or in addition to HER
inhibitors, such as trastuzumab or lapatinib, in various types of
breast cancer (88). The clinical
evaluation of MK-2206 for the treatment of acute myelogenous
leukemia proved to be largely unsatisfactory (89). The combination of MK-2206 and
selumetinib, a MEK1/2 inhibitor, did not achieved the desired level
of clinical activity in patients with advanced colorectal cancer
(90).
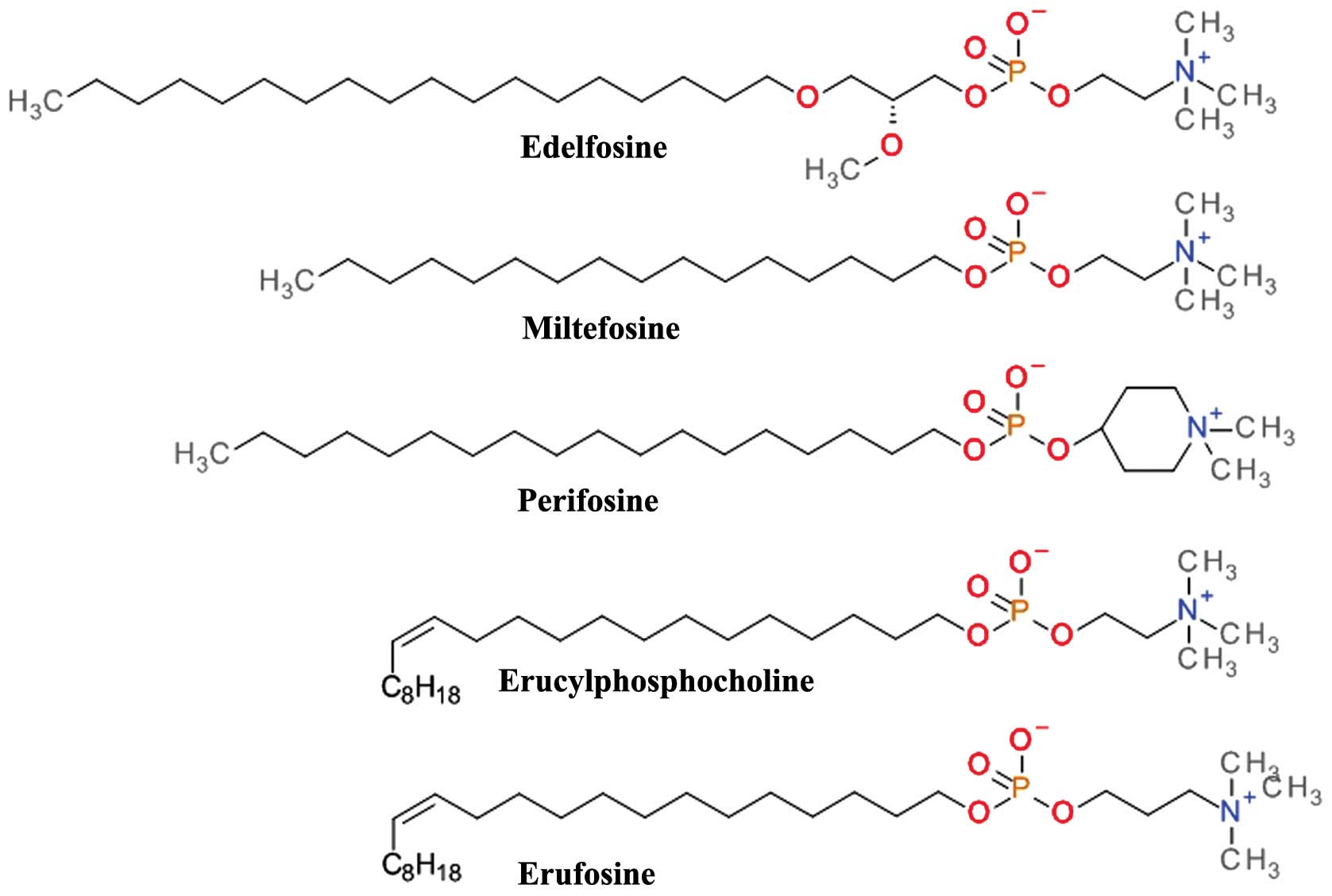
Alkylphospholipids (ALPs)
Structurally based on the scaffold of
lysophosphatidylcholine, ALPs have a long hydrocarbon chain that
allows easy partitioning into the plasma membrane of cells and thus
accumulation into cell membranes (91). They kill tumor cells by the
induction apoptotic and non-apoptotic cell death, and indirectly by
interference with the PH domain of Akt and disrupting critical
signal transduction pathways (92). ALPs (Fig. 7) prevent plasma membrane
recruitment of the PH domain of Akt by disrupting membrane
microdomains and/or by displacing its natural ligands from the PH
domain. The result is that Akt is no longer capable of adopting the
favorable conformation for its phosphorylation and activation
(93).
Edelfosine
(1-O-octadecy-2-O-methyl-rac-glycero-3-phosphocholine,
Et-18-OCH3) is one of the first synthetic analogues of
lysophosphatidylcholine (94)
which has shown promising anti-proliferative effects in both in
vivo and in vitro models. Cells treated with edelfosine
exhibited a marked and rapid decrease in p-Akt Ser473 levels,
coupled with a reduction in the phosphorylation levels of mTOR
(p-mTOR) (95). Its clinical use
is limited by the high toxicity and low selectivity (96). The thioether analog of edelfosine,
ilmofosine (BM 41.440) has demonstrated dose-dependent in
vivo and in vitro antitumor activity in various solid
tumor models. The gastrointestinal toxicity is dose-limiting and
limits its clinical use (97).
The glycerol moiety in ALP has proven to be not
essential for the antitumor activity and a second generation
containing a phosphoester chain was designed and synthesized.
Miltefosine (hexadecylphosphocholine, HePC) was evaluated asan oral
therapy in clinical studies against soft tissue sarcomas (98) and advanced colorectal cancer
(99); however, the doses required
for the antitumor effects were too toxic. Miltefosine, either used
alone or in conjunction with other therapies, proved to be
effective and tolerable as a local treatment for cutaneous breast
cancer (100). The clinical use
of miltefosine is restricted to topical application due to
hemolytic toxicity upon intravenous application. Miltefosine has
demonstrated very good activity against various parasite species
and is one of the few therapeutic solutions for visceral and
cutaneous leishmaniasis (101).
The replacement of the choline moiety of miltefosine
with a piperidine scaffold resulted in D-21266 (perifosine), a
compound with a better metabolic stability, and a significantly
improved gastrointestinal tract tolerance. Perifosine has displayed
significant anti-proliferative activity and triggers apoptosis
in vitro and in vivo in several human tumor model
systems. Perifosine has been tested in several clinical trials
against a large variety of tumors. As a single therapy, perifosine
proved useful only in sarcoma and Waldenstrom macroglobulinemia. It
is currently in clinical development for the treatment of
colorectal cancer in combination with capecitabine and of multiple
myeloma in combination with bortezomib and dexamethasone or in
combination with lenalidomide and dexamethasone (102). Perifosine has been tested in
combination with sorafenib, sunitinib, paclitaxel, docetaxel,
lenalinomide or gemcitabine in various other types of cancer
(91).
Erucylphosphocholine (ErPC) is a long chain and
unsaturated homologue of miltefosine which causes it to associate
in aqueous environments as non-haemolytic lamellar rather than
micellar structures making it suitable for intravenous
administration (103). Its
clinical use is difficult due to the poor solubility in aqueous
solutions (93). The search for
structural analogues with improved solubility properties resulted
in Erufosine (ErPC3, erucylphosphohomocholine) (103). Both ErPC and ErPC3 are superior
to other ALPs in their ability to cross the blood-brain barrier and
accumulate in brain tissue and constitute promising candidates for
glioblastoma therapy (104).
ErPC and ErPC3 have been tested in various other
cancer types of cancer, including acute myeloid leukemia, chronic
lymphocytic leukemia, colorectal cancer and oral squamous cell
carcinoma (105). Preclinical
data suggest that ErPC and ErPC3 have additive or synergistic
effects when combined with ionizing radiation and may be used with
radiation therapy to overcome resistance to standard treatment
(104,106).
Indole-3-carbinol and analogues
Indole-3-carbinol is a natural compound found in the
Brassica species with average anti-proliferative effects and
chemopreventive activity against chemically-induced tumors in
various rodent models through the inactivation of Akt kinase
(107). The main disadvantage of
indole-3-carbinol is its low chemical stability, under acidic
conditions being converted to 3,3′-diindolylmethane which is
considered responsible for its biological effects in vivo
(108).
In pursuit of compounds with better stability,
3-chloroacetylindole was developed as a potent ATP non-competitive
inhibitor of Akt1 and Akt2, and has been shown to suppress cell
growth and induce apoptosis both in vitro and in vivo
experiments (109).
Based on the structure of diindolylmethane, a novel
class of indole analogs were developed in order to optimize its
anticancer effects. The most promising of these analogs, an
indolo[2,3-b]carbazole derivative, has exhibited potent oral
anticancer activity against various types of cancer and no
significant toxicity (110).
SR13668 has been shown to have no adverse effects on fasting
glucose levels or body weight in mice treated with doses of up to
500 mg/kg for 14 days (110).
Preclinical pharmacokinetic studies found low bioavailability in
rats, dogs and monkeys due to the limited aqueous solubility and
expected high permeability through biological membranes (111). SR13668 was investigated in a
clinical trial on healthy volunteers to assess the formulation
effect on the bioavailability after oral administration (112).
OSU-A9 is an indole-3-carbinol derivative with
better chemical stability that has been shown to inhibit Akt
signaling in MCF-7 cells, as demonstrated by the concomitant
dephosphorylation of Akt and two downstream kinase substrates,
GSK-3β and IKKα. This new derivative was also found to inhibit the
nuclear factor-κB signaling pathways (113).
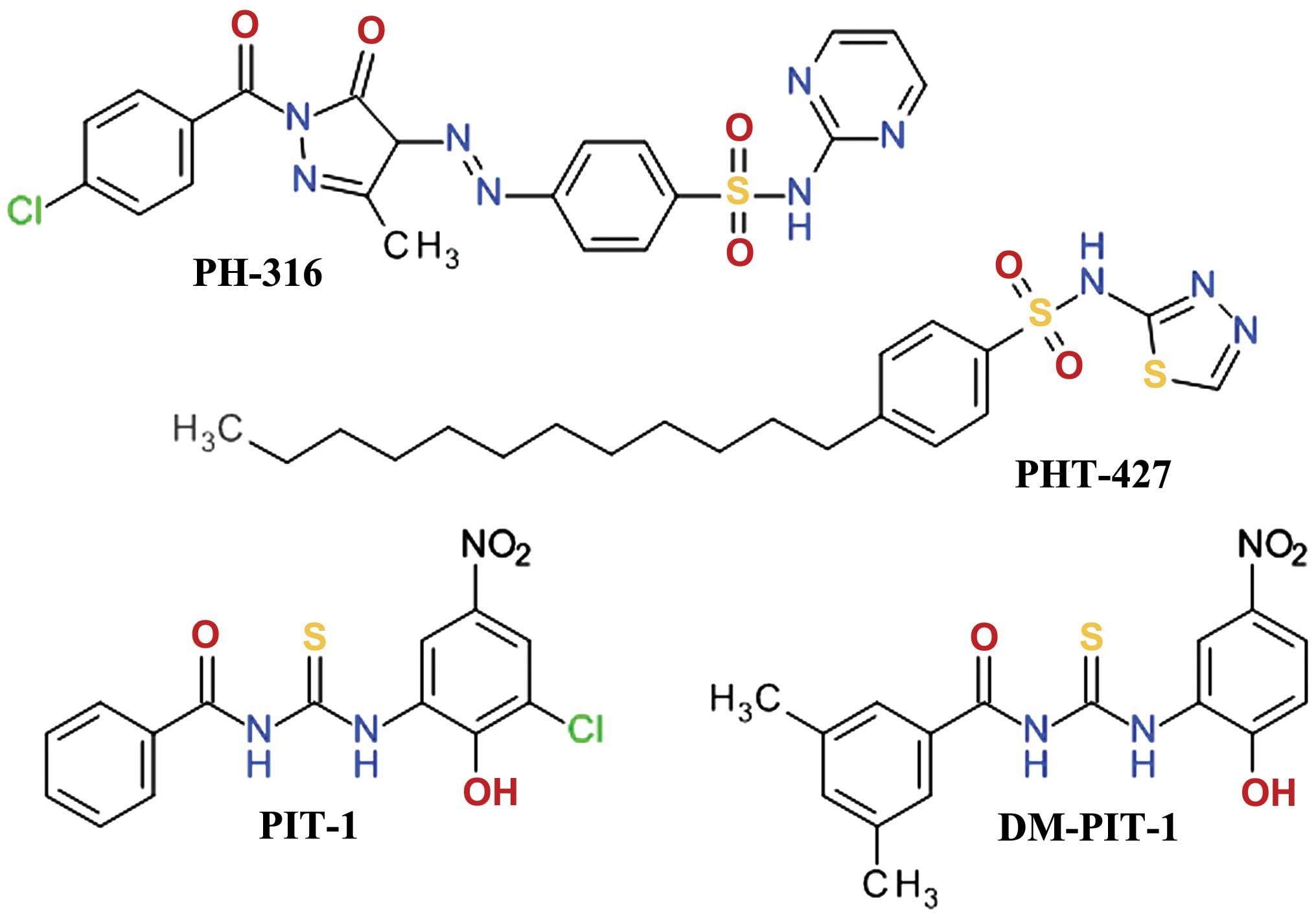
Sulfonamide derivatives
A three-dimensional virtual screening based on the
Akt1 PH domain structure led to the development of compound PH-316,
a sulfadiazine derivative (Fig.
8), which inhibited Akt and HT-29 cell proliferation. However,
it failed to achieve the blood concentration required to inhibit
Akt in animal models, due to the rapid metabolism of the azo bond
(114). In order to improve the
anticancer profile of PH-316, a larger in silico study was
performed and four scaffolds were identified. Several structural
modifications led to PHT-427
(4-dodecyl-N-(1,3,4-thiadiazol-2-yl) benzenesulfonamide), a
potent Akt inhibitor with an IC50 of 6.3±0.9 μmol/l in
Panc-1 cells and a strong inducer of apoptosis at 20 μmol/l
(115). PHT-427 inhibited the
growth of various human tumor xenografts in immunodeficient mice
following oral administration. It showed synergic antitumor
activity with paclitaxel and gemcitabine in a Panc-1 tumors, with
erlotinib in a non-small-cell lung cancers and with paclitaxel in
breast cancer (116).
Thiourea derivatives
High-throughput fluorescence polarization binding
assay based on a recombinant fragment of human Akt1 identified two
inhibitors of PIP3/PH domain binding, which were termed PIT-1 and
PIT-2. They also inhibited, although with lower affinity, the
binding of PIP3 to the PH domains of PDK1, general receptor for
phosphoinositides isoform 1 (GRP1) and ARNO (117). Structural analysis demonstrated
that thiourea and hydroxyl groups are critical for the anticancer
activity. Due to the limited aqueous solubility of PIT-1, the
dimethyl analog, DM-PIT-1 (Fig.
8), was developed (118). In
a study focusing on the synthesis of new pyrazole derivatives based
on the tenovins scaffold, we obtained
N-[(1-methyl-1H-pyrazol-4-yl)
carbonyl]-N′-(3-bromophenyl)-thiourea as an apoptosis agent and
biological screening suggested that it can inhibit Akt in a similar
manner with PIT-1 (119).
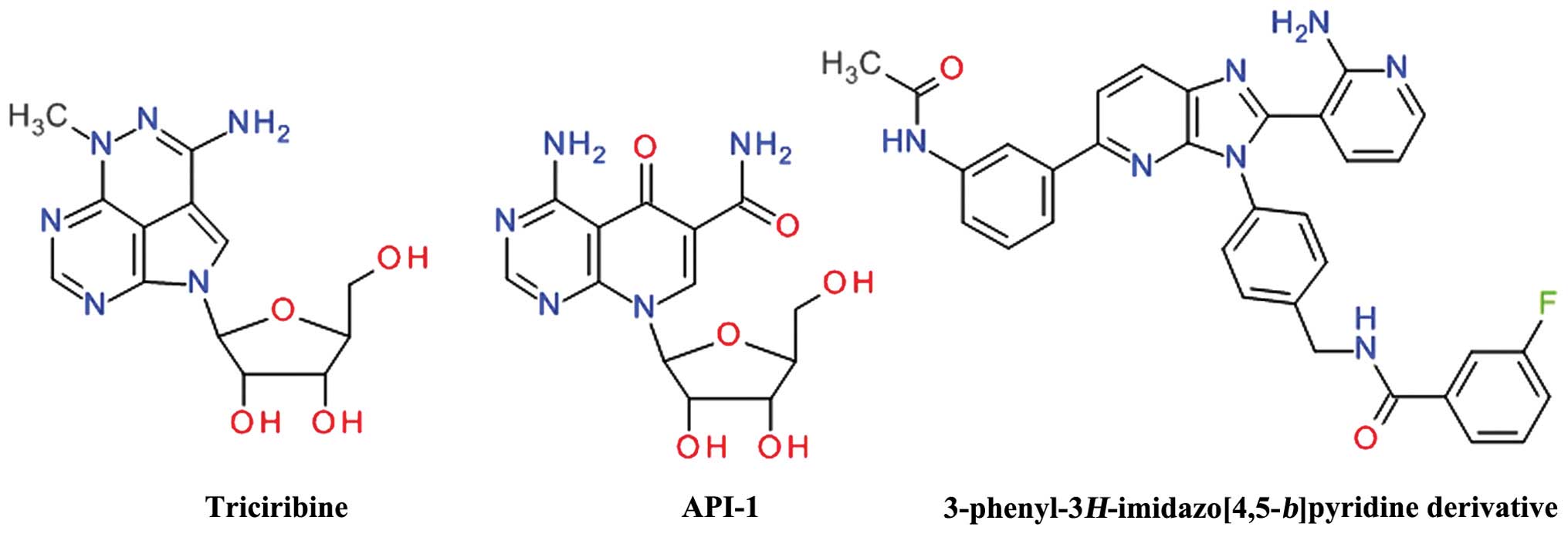
Purine derivatives
Screening of the National Cancer Institute (NCI)
Diversity Set led to the identification of API-2 [(Triciribine
(TCN), NSC 154020] that potently inhibited Akt signaling in human
cancer cells, leading to the inhibition of cell growth and the
induction of apoptosis (120).
TCN is a tricyclic purine nucleoside derivative (Fig. 9) that is metabolically activated
inside cells by adenosine kinase to its monophosphate active
analog, TCN-P (121). Surface
plasmon resonance and nuclear magnetic resonance spectroscopy have
demonstrated that TCN-P, but not TCN, binds to the PH domain of Akt
in the vicinity of the PIP3 binding pocket, thereby preventing Akt
phosphorylation and subsequent activation. The proposed mechanism
of TCN-P Akt inhibition is by preventing PIP3 from recruiting Akt
to the plasma membrane, either by competing with PIP3 for binding
to PH domain or by binding to region that induces conformational
changes that hinder PIP3 activation (122). Treatment of T cell acute
lymphocytic leukemia cells with 10 μM TCN has been shown to inhibit
Akt phosphorylation and its downstream signaling, causing cell
cycle arrest and caspase-dependent apoptosis (123). In prostate cancer cells, TCN was
shown to increase the apoptosis induced by the death receptor
pathway (124). TCN and TCN-P
were subjected to several clinical trials against various solid
neoplasms and hematological malignancies, but their efficacy is
limited due to their toxicity (121). The combination treatment of TCN
with other anticancer agents proved to be a better solution than
treatment with TCN alone (125).
An isosteric scaffold of adenosine, the
4-aminopyrido[2,3-d]pyrimidine derivative API-1 was
discovered to inhibit Akt by screening the DTP/NCI compound library
in a cell-based assay. API-1 binds to the PH domain and inhibits
Akt membrane translocation, leading to the inhibition of the growth
of tumors with hyperactivated Akt (126). API-1 was shown to induce
apoptosis in tested cancer cell lines by synergizing with
TNF-related apoptosis-inducing ligand (TRAIL) (127). A series of
4-amino-pyrrolo[2,3-d]pyrimidine derivatives, closely
related to API-1, were discovered in a high-throughput screening
based on a newly developed fluorescence-based assay as allosteric
Akt inhibitors (85).
A series of
3-phenyl-3H-imidazo[4,5-b]pyridine was developed as
orally bioavailable potent ATP non-competitive Akt inhibitors, the
best compound being the
N-(4-(5-(3-acetamidophenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]
pyridin-3-yl)benzyl)-3-fluorobenzamide (128). Even if the
imidazo[4,5-b]pyridine template is closely related to the
purine ring, the structural conformation of these compounds
resemble more the 2,3-diphenylquinoxalines based derivatives.
ARQ 092 was identified searching for inhibitors
which use the intrinsic negative regulatory function of hydrophobic
clusters in the ATP-binding cleft. ARQ 092 binds to inactive,
unphosphorylated Akt1 with subnanomolar affinity and inhibits all
three isoforms. In a previous study, the growth of AN3CA human
endometrial tumors was markedly suppressed in xenograft models
following the oral administration of ARQ 092 (129). ARQ 092 and its congener, ARQ 751,
have been shown to inhibit proliferation across multiple tumor
types and were most potent in leukemia, breast, endometrial and
colorectal cancer cell lines (130). Only the core structure of these
compounds was disclosed and consists of
3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines.
The authors do not mention the developmental process, but most
probably it involved the aforementioned
N-(4-(5-(3-acetamidophenyl)-2-(2-aminopyridin-3-yl)-3H-imidazo[4,5-b]pyridin-3-yl)benzyl)-3-fluorobenzamide.
Derivatives with various structures
BAY 1125976 has been reported as a highly
selective, potent allosteric Akt1/2 inhibitor, with strong in
vitro and in vivo activity in tumor models with
activated Akt signaling and in cell lines possessing the activating
mutation, Akt1E17K (131). The
structure of the compound was not yet disclosed.
Using in silico screening, derivatives of
3-methylxanthine, quinoline-4-carboxamide and
2-[4-(cyclohexa-1,3-dien-1-yl)-1H-pyrazol-3-yl]phenol were
proposed as potential leads for future development of Akt1
allosteric inhibitors (132).
A series of tetracyclic triterpenoids isolated from
the oleogum resin of Boswellia carterii were discovered as
potent Akt inhibitors, whereas, 3-oxo-tirucallic acid is a weak
inhibitor of Akt, 3α-.and 3β-acetoxy-tirucallic acids block Akt1
activity at concentrations below 1 μmol/l. Treatment of PC-3 cells
with acetoxy-tirucallic acids was shown to inhibit the
phosphorylation of GSK-3β and reduce the nuclear levels of
β-catenin and c-Myc. Molecular docking studies indicated that
tirucallic acids exhibit affinity binding to the PH domain of Akt.
Treatment of mice bearing xenotransplanted PC-3 tumors for 2 weeks
with 10 μmol/kg 3β-acetoxy-tirucallic resulted in a significant
reduction in tumor growth (133).
6. Irreversible inhibitors
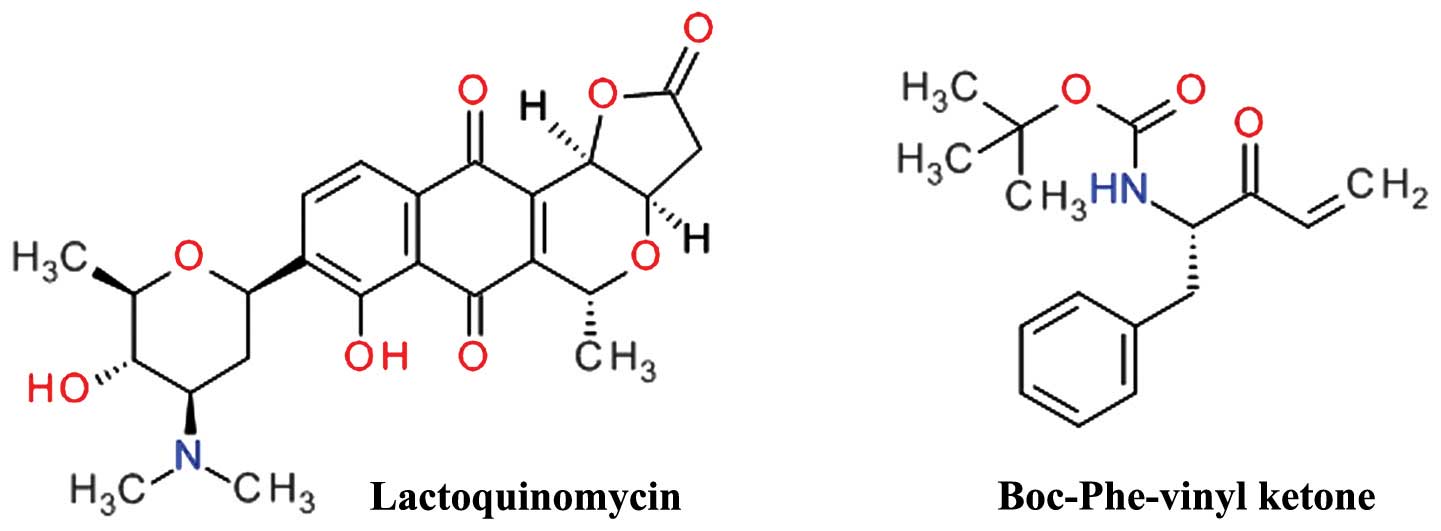
Lactoquinomycin is a natural product isolated from
the fermentation broth of Streptomyces sp. LL-AF101 that was
found to selectively inhibit Akt (134). Lactoquinomycin is a
pyrano-naphthoquinone antibiotic (Fig. 10) and a potent inhibitor of Akt by
irreversible covalent interaction with two critical catalytic
activation loop cysteines (Cys296 and Cys310) and does not
interfere with the PDK1-dependent Thr308 phosphorylation (135). The proposed mechanism is a
S-alkylation involving the reduction of the quinone ring to a
hydroquinone and subsequent opening of the lactone ring (136). In various tumor cell models,
lactoquinomycin potently inhibited the phosphorylation of Akt
downstream substrates GSK-3α/β, FKHRL1 and mTOR (135). Frenolicin B is a related
pyrano-naphthoquinone natural derivative with similar Akt
inhibition profile (136). A
similar inhibitory effect was observed for kalafungin and
medermycin (136).
A phenylalanine vinyl ketone derivative (Fig. 10) was designed and synthesized as
a covalent inactivator of Akt, and demonstrated potent cell growth
inhibition in HCT116 and H460 cells and selective Akt inhibition by
bonding to Cys310 (137).
Alkylation of Cys310 by Michael acceptors was demonstrated also for
4-hydroxynonenal (138).
The 1,6-naphthyridinone and imidazo-1,2-pyridine
templates were used to design Michael acceptors to target Cys296 or
Cys310. The lead compound exhibited a selective profile,
exclusively targeting Akt isoforms without affecting related
kinases (139).
7. Clinically relevant Akt inhibitors
Despite the large number of compounds proven to
inhibit Akt in vitro and in vivo models, only a
limited number of these have entered clinical evaluation.
Miltefosine is the only Akt inhibitor approved, but only for the
treatment of visceral and cutaneous leishmaniasis, and not for the
treatment of cancer. The compounds evaluated clinically and the
status of the trials are summarized in Table II.
| Table IIReported clinical evaluations of Akt
inhibitors. |
Table II
Reported clinical evaluations of Akt
inhibitors.
| Therapeutic
regimen | Indication | Status |
|---|
| GSK690693 | Hematological
neoplasia, acute lymphoblastic leukemia | Clinical
development terminated due to hyperglycemia |
| AZD5363
monotherapy | Breast cancer,
gastric cancer, prostate cancer | Phase I/phase II
clinical trials |
| Afuresertib
(GSK2110183) monotherapy | Relapsed or
refractory multiple myeloma | Phase I/phase II
clinical trials |
| Afuresertib
(GSK2110183) in combination with bortezomib or dexamethasone | Relapsed or
refractory multiple myeloma | Phase I/Phase II
clinical trials |
| Uprosertib
(GSK2141795) monotherapy | Relapsed or
refractory multiple myeloma | Phase I/phase II
clinical trials |
| Uprosertib
(GSK2141795) in combination with trametinib | Relapsed or
refractory multiple myeloma | Phase I/phase II
clinical trials |
| Ipatasertib
(GDC-0068, RG7440) monotherapy | Triple-negative
breast cancer | Phase I/phase II
clinical trials |
| MK-2206 in
combination with gefitinib or erlotinib | Advanced
non-small-cell lung carcinoma | Phase I/phase II
clinical trials |
| MK-2206
monotherapy | Acute myelogenous
leukemia | Unsatisfactory
clinical results
Usage limited to topical application due to hemolytic toxicity
after intravenous injection |
| MK-2206 in
combination with selumetinib | Colorectal
carcinoma | Unsatisfactory
clinical results
Similar toxicity as for monotherapy |
| MK-2206
monotherapy | Non-cancerous
disease (visceral and cutaneous leishmaniasis) | Early clinical
evaluation |
| TCN or TCN-P
monotherapy | Solid tumors,
hematological malignancies | Clinical efficacy
limited due to toxicity |
Based on the cellular function of Akt and on the
preclinical studies of the efficacy of various Akt inhibitors, the
strategy of combined therapy emerged as a major research direction.
On the other hand, Akt inhibitors have been shown to significantly
improve the therapeutic outcome of traditional anticancer drugs.
The preclinical tests of several Akt inhibitors as combinative
therapy are summarized in Table
III.
| Table IIIReported preclinical combinations
with other therapeutic modalities. |
Table III
Reported preclinical combinations
with other therapeutic modalities.
| Akt inhibitor | Combination
with | Action |
|---|
| A-674563,
A-443654 | Microtubule
inhibitor (paclitaxel) | Inhibition of Akt
through PKA |
| Afuresertib
(GSK2110183) | Proteasome
inhibitor (bortezomib)
Anti-inflammatory drug (dexamethasone) | Inhibition of Akt1,
Akt2, Akt3 |
| Uprosertib
(GSK2141795) | MEK1, MEK2 kinase
inhibitor (trametinib) | Inhibition of Akt1,
Akt2, Akt3 |
| MK-2206 | Topoisomerase
inhibitors (doxorubicin, camptothecin)
Nucleotide analogues (gemcitabine, 5-FU)
DNA cross-linker (carboplatin)
EGFR inhibitors (erlotinib, gefitinib, ZD1839,
trastuzumab)
Kinase inhibitors (lapatinib, selumetinib) | G1 arrest,
apoptosis induction |
| Perifosine
(D-21266) | Capecitabine
(prodrug of nucleotide analogue 5-FU) | Inhibition of
p-mTOR, apoptosis induction |
|
Erucylphosphocholine (ErPC), erufosine
(ErPC3, erucylphosphohomocholine) | Radiation
therapy | Inhibition of
p-mTOR |
| TCN, TCN-P | Nucleotide analogue
(gemcitabine) | Inhibition of
p-Akt |
8. Potential biomarker for Akt
inhibitors
In the era of precision medicine identifying
predictive biomarkers, therapy with targeted agents has become a
major goal of translational research. Therefore, potential
biomarkers for treatment with Akt inhibitors would facilitate the
design of clinical trials and would accelerate the introduction of
these compounds to clinical practice. In this regard, the
observation that sensitivity to Akt-specific inhibitors is
dependent upon the activation of the PI3K/Akt/mTOR pathway suggests
that biomarkers in this pathway may by predictive for treatment
with Akt inhibitors (140).
Similarly, experiments using cell lines with PIK3CA or PTEN
alterations have demonstrated increased activity for all MK-2206,
AZD5363 and GDC-0068 compounds (63,141,142).
In the clinic, early-phase trials with MK-2206 and
ipatasertib support the hypothesis that AKT inhibitors do possess
activity against tumors with PTEN deficiency. Objective responses
have been reported in patients with PTEN-deficient metastatic
pancreatic and colorectal cancer (143,144). In addition to the loss of PTEN,
clinical evidence suggests that patients with PIK3CA-mutant tumors
may benefit from treatment with Akt inhibitors. For example, in
phase I trials of single-agent ipatasertib and AZD5363, objective
response and clinical benefit were observed in patients with
PIK3CA-mutant colorectal cancer (145), as well as gynecological
malignancies (cervical and endometrial cancer) with PIK3CA
mutations (146). In conclusion,
analysis for somatic alteration in PTEN or PIK3CA mutation may
serve as a potent biomarker to therapy with Akt inhibitors.
9. Conclusions
Akt modulates and regulates the function of
numerous substrates and is the key factor in cell survival and
growth mechanism. A plethora of studies supports the role of Akt as
a well validated target for drug development. In spite of modern
drug discovery techniques and increasing knowledge regarding Akt
functions and activation, no Akt inhibitor has been yet approved
for oncologic use. This is further coupled with a relatively low
number of compounds that have been included in clinical trials.
The drug discovery process is hindered by a high
degree of structural similarity between Akt isoforms, especially in
the catalytic domain, and their considerable structural analogy to
the AGC kinase family. The use of alternative approaches employing
allosteric inhibitors of Akt produced compounds with better
selectivity and efficacy.
All the major components of the Akt pathway,
consisting of PI3Ks, PDK1, Akt and mTOR, are the focus of intensive
research for targeted cancer therapy (32). However, thus far a limited number
of drugs has emerged from this approach.
Clinical trials with MK-2206 and GDC-0068 support
the hypothesis that Akt inhibitors will be most useful in tumors
with PTEN deficiency and PIK3 mutations (147). However, most Akt inhibitors
display a rather limited clinical activity as single agents. This
clearly emphasizes the importance of combinatorial treatments for
better therapeutic perspective of Akt inhibitors.
Acknowledgements
The authors acknowledge the financial support
offered by Romanian National Authority for Scientific Research,
UEFISCDI, through grant PN-II-PCCA-2 no. 136/2012 and grant
PN-II-RU-TE-2014-4-1670, no. 342/2015.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steelman LS, Stadelman KM, Chappell WH,
Horn S, Bäsecke J, Cervello M, Nicoletti F, Libra M, Stivala F,
Martelli AM, et al: Akt as a therapeutic target in cancer. Expert
Opin Ther Targets. 12:1139–1165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bellacosa A, Kumar CC, Di Cristofano A and
Testa JR: Activation of AKT kinases in cancer: Implications for
therapeutic targeting. Adv Cancer Res. 94:29–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arcaro A and Guerreiro AS: The
phosphoinositide 3-kinase pathway in human cancer: Genetic
alterations and therapeutic implications. Curr Genomics. 8:271–306.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitsiades CS, Mitsiades N and Koutsilieris
M: The Akt pathway: Molecular targets for anti-cancer drug
development. Curr Cancer Drug Targets. 4:235–256. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martelli AM, Tabellini G, Bressanin D,
Ognibene A, Goto K, Cocco L and Evangelisti C: The emerging
multiple roles of nuclear Akt. Biochim Biophys Acta - Mol Cell Res.
1823.2168–2178. 2012.
|
|
10
|
Arencibia JM, Pastor-Flores D, Bauer AF,
Schulze JO and Biondi RM: AGC protein kinases: From structural
mechanism of regulation to allosteric drug development for the
treatment of human diseases. Biochim Biophys Acta. 1834.1302–1321.
2013.
|
|
11
|
Davis WJ, Lehmann PZ and Li W: Nuclear
PI3K signaling in cell growth and tumorigenesis. Front Cell Dev
Biol. 3:242015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sasaki T, Yamashita Y and Kuniyasu H: AKT
plays a crucial role in gastric cancer (Review). Oncol Lett.
10:607–611. 2015.PubMed/NCBI
|
|
13
|
Shayesteh L, Lu Y, Kuo W-L, Baldocchi R,
Godfrey T, Collins C, Pinkel D, Powell B, Mills GB and Gray JW:
PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet.
21:99–102. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine DA, Bogomolniy F, Yee CJ, Lash A,
Barakat RR, Borgen PI and Boyd J: Frequent mutation of the PIK3CA
gene in ovarian and breast cancers. Clin Cancer Res. 11:2875–2878.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Andjelković M, Alessi DR, Meier R,
Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM and
Hemmings BA: Role of translocation in the activation and function
of protein kinase B. J Biol Chem. 272:31515–31524. 1997. View Article : Google Scholar
|
|
16
|
Carnero A and Paramio JM: The
PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front Oncol.
4:2522014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Conus NM, Hannan KM, Cristiano BE,
Hemmings BA and Pearson RB: Direct identification of tyrosine 474
as a regulatory phosphorylation site for the Akt protein kinase. J
Biol Chem. 277:38021–38028. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Legate KR, Montañez E, Kudlacek O and
Fässler R: ILK, PINCH and parvin: The tIPP of integrin signalling.
Nat Rev Mol Cell Biol. 7:20–31. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jhaveri K and Modi S: Ganetespib: Research
and clinical development. Onco Targets Ther. 8:1849–1858.
2015.PubMed/NCBI
|
|
20
|
O‘Neill AK, Niederst MJ and Newton AC:
Suppression of survival signalling pathways by the phosphatase
PHLPP. FEBS J. 280:572–583. 2013. View Article : Google Scholar :
|
|
21
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Altomare DA and Khaled AR: Homeostasis and
the importance for a balance between AKT/mTOR activity and
intracellular signaling. Curr Med Chem. 19:3748–3762. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cohen MM Jr: The AKT genes and their roles
in various disorders. Am J Med Genet A. 161A:2931–2937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Emamian ES: AKT/GSK3 signaling pathway and
schizophrenia. Front Mol Neurosci. 5:332012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mackenzie RWA and Elliott BT: Akt/PKB
activation and insulin signaling: A novel insulin signaling pathway
in the treatment of type 2 diabetes. Diabetes Metab Syndr Obes.
7:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Osorio-Fuentealba C and Klip A: Dissecting
signalling by individual Akt/PKB isoforms, three steps at once.
Biochem J. 470:e13–e16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Easton RM, Cho H, Roovers K, Shineman DW,
Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T,
et al: Role for Akt3/protein kinase Bgamma in attainment of normal
brain size. Mol Cell Biol. 25:1869–1878. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
West KA, Castillo SS and Dennis PA:
Activation of the PI3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar
|
|
29
|
Candido S, Rapisarda V, Marconi A,
Malaponte G, Bevelacqua V, Gangemi P, Scalisi A, McCubrey JA,
Maestro R, Spandidos DA, et al: Analysis of the
B-RafV600E mutation in cutaneous melanoma patients with
occupational sun exposure. Oncol Rep. 31:1079–1082. 2014.PubMed/NCBI
|
|
30
|
Shi Y, Liu X, Han EK, Guan R, Shoemaker
AR, Oleksijew A, Woods KW, Fisher JP, Klinghofer V, Lasko L, et al:
Optimal classes of chemotherapeutic agents sensitized by specific
small-molecule inhibitors of akt in vitro and in vivo. Neoplasia.
7:992–1000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hafsi S, Pezzino FM, Candido S, Ligresti
G, Spandidos DA, Soua Z, McCubrey JA, Travali S and Libra M: Gene
alterations in the PI3K/PTEN/AKT pathway as a mechanism of
drug-resistance (Review). Int J Oncol. 40:639–644. 2012.
|
|
32
|
Carnero A: The PKB/AKT pathway in cancer.
Curr Pharm Des. 16:34–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva
FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al: PTEN
activation contributes to tumor inhibition by trastuzumab, and loss
of PTEN predicts trastuzumab resistance in patients. Cancer Cell.
6:117–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fujita T, Doihara H, Washio K, Kawasaki K,
Takabatake D, Takahashi H, Tsukuda K, Ogasawara Y and Shimizu N:
Proteasome inhibitor bortezomib increases PTEN expression and
enhances trastuzumab-induced growth inhibition in
trastuzumab-resistant cells. Anticancer Drugs. 17:455–462. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Olaru OT, Niţulescu GM, Orţan A and
Dinu-Pîrvu CE: Ethnomedicinal, Phytochemical and Pharmacological
Profile of Anthriscus sylvestris as an Alternative Source for
Anticancer Lignans. Molecules. 20:15003–15022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peuhu E, Rivero-Müller A, Stykki H,
Torvaldson E, Holmbom T, Eklund P, Unkila M, Sjöholm R and Eriksson
JE: Inhibition of Akt signaling by the lignan matairesinol
sensitizes prostate cancer cells to TRAIL-induced apoptosis.
Oncogene. 29:898–908. 2010. View Article : Google Scholar
|
|
37
|
Shoji K, Oda K, Nakagawa S, Hosokawa S,
Nagae G, Uehara Y, Sone K, Miyamoto Y, Hiraike H, Hiraike-Wada O,
et al: The oncogenic mutation in the pleckstrin homology domain of
AKT1 in endometrial carcinomas. Br J Cancer. 101:145–148. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rychahou PG, Kang J, Gulhati P, Doan HQ,
Chen LA, Xiao SY, Chung DH and Evers BM: Akt2 overexpression plays
a critical role in the establishment of colorectal cancer
metastasis. Proc Natl Acad Sci USA. 105:20315–20320. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Graff JR, Konicek BW, McNulty AM, Wang Z,
Houck K, Allen S, Paul JD, Hbaiu A, Goode RG, Sandusky GE, et al:
Increased AKT activity contributes to prostate cancer progression
by dramatically accelerating prostate tumor growth and diminishing
p27Kip1 expression. J Biol Chem. 275:24500–24505. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Altomare DA, Tanno S, De Rienzo A,
Klein-Szanto AJ, Tanno S, Skele KL, Hoffman JP and Testa JR:
Frequent activation of AKT2 kinase in human pancreatic carcinomas.
J Cell Biochem. 87:470–476. 2002. View Article : Google Scholar
|
|
41
|
Lin HP, Lin CY, Huo C, Jan YJ, Tseng JC,
Jiang SS, Kuo YY, Chen SC, Wang CT, Chan TM, et al: AKT3 promotes
prostate cancer proliferation cells through regulation of Akt,
B-Raf, and TSC1/TSC2. Oncotarget. 6:27097–27112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cristiano BE, Chan JC, Hannan KM, Lundie
NA, Marmy-Conus NJ, Campbell IG, Phillips WA, Robbie M, Hannan RD
and Pearson RB: A specific role for AKT3 in the genesis of ovarian
cancer through modulation of G(2)-M phase transition. Cancer Res.
66:11718–11725. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mattmann ME, Stoops SL and Lindsley CW:
Inhibition of Akt with small molecules and biologics: Historical
perspective and current status of the patent landscape. Expert Opin
Ther Pat. 21:1309–1338. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kumar CC and Madison V: AKT crystal
structure and AKT-specific inhibitors. Oncogene. 24:7493–7501.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Böckmann S and Nebe B: The in vitro
effects of H-89, a specific inhibitor of protein kinase A, in the
human colonic carcinoma cell line Caco-2. Eur J Cancer Prev.
12:469–478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Reuveni H, Livnah N, Geiger T, Klein S,
Ohne O, Cohen I, Benhar M, Gellerman G and Levitzki A: Toward a PKB
inhibitor: Modification of a selective PKA inhibitor by rational
design. Biochemistry. 41:10304–10314. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Breitenlechner CB, Wegge T, Berillon L,
Graul K, Marzenell K, Friebe WG, Thomas U, Schumacher R, Huber R,
Engh RA, et al: Structure-based optimization of novel azepane
derivatives as PKB inhibitors. J Med Chem. 47:1375–1390. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Murray AJ: Pharmacological PKA inhibition:
All may not be what it seems. Sci Signal. 1:re4-re42008. View Article : Google Scholar
|
|
49
|
Heerding DA, Rhodes N, Leber JD, Clark TJ,
Keenan RM, Lafrance LV, Li M, Safonov IG, Takata DT, Venslavsky JW,
et al: Identification of
4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol
(GSK690693), a novel inhibitor of AKT kinase. J Med Chem.
51:5663–5679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Levy DS, Kahana JA and Kumar R: AKT
inhibitor, GSK690693, induces growth inhibition and apoptosis in
acute lymphoblastic leukemia cell lines. Blood. 113:1723–1729.
2009. View Article : Google Scholar
|
|
51
|
Carol H, Morton CL, Gorlick R, Kolb EA,
Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C, Smith MA, et
al: Initial testing (stage 1) of the Akt inhibitor GSK690693 by the
pediatric preclinical testing program. Pediatr Blood Cancer.
55:1329–1337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Crouthamel MC, Kahana JA, Korenchuk S,
Zhang SY, Sundaresan G, Eberwein DJ, Brown KK and Kumar R:
Mechanism and management of AKT inhibitor-induced hyperglycemia.
Clin Cancer Res. 15:217–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Donald A, McHardy T, Rowlands MG, Hunter
LJ, Davies TG, Berdini V, Boyle RG, Aherne GW, Garrett MD and
Collins I: Rapid evolution of 6-phenylpurine inhibitors of protein
kinase B through structure-based design. J Med Chem. 50:2289–2292.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Caldwell JJ, Davies TG, Donald A, McHardy
T, Rowlands MG, Aherne GW, Hunter LK, Taylor K, Ruddle R, Raynaud
FI, et al: Identification of
4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d] pyrimidines as
selective inhibitors of protein kinase B through fragment
elaboration. J Med Chem. 51:2147–2157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yap TA, Walton MI, Hunter L-JK, Valenti M,
de Haven Brandon A, Eve PD, Ruddle R, Heaton SP, Henley A, Pickard
L, et al: Preclinical pharmacology, antitumor activity, and
development of pharmacodynamic markers for the novel, potent AKT
inhibitor CCT128930. Mol Cancer Ther. 10:360–371. 2011. View Article : Google Scholar
|
|
56
|
Freeman-Cook KD, Autry C, Borzillo G,
Gordon D, Barbacci-Tobin E, Bernardo V, Briere D, Clark T, Corbett
M, Jakubczak J, et al: Design of selective, ATP-competitive
inhibitors of Akt. J Med Chem. 53:4615–4622. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lippa B, Pan G, Corbett M, Li C, Kauffman
GS, Pandit J, Robinson S, Wei L, Kozina E, Marr ES, et al:
Synthesis and structure based optimization of novel Akt inhibitors.
Bioorg Med Chem Lett. 18:3359–3363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
McHardy T, Caldwell JJ, Cheung KM, Hunter
LJ, Taylor K, Rowlands M, Ruddle R, Henley A, de Haven Brandon A,
Valenti M, et al: Discovery of 4-amino-1-(7H-pyrrolo[2,3-d]
pyrimidin-4-yl)piperidine-4-carboxamides as selective, orally
active inhibitors of protein kinase B (Akt). J Med Chem.
53:2239–2249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Addie M, Ballard P, Buttar D, et al:
Discovery of
4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]
pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an orally
bioavailable, potent inhibitor of Akt kinases. J Med Chem.
56:2059–2073. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Crafter C, Vincent JP, Tang E, Dudley P,
James NH, Klinowska T and Davies BR: Combining AZD8931, a novel
EGFR/HER2/HER3 signalling inhibitor, with AZD5363 limits AKT
inhibitor induced feedback and enhances antitumour efficacy in
HER2-amplified breast cancer models. Int J Oncol. 47:446–454.
2015.PubMed/NCBI
|
|
61
|
Lamoureux F and Zoubeidi A: Dual
inhibition of autophagy and the AKT pathway in prostate cancer.
Autophagy. 9:1119–1120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Blake JF, Xu R, Bencsik JR, Xiao D, Kallan
NC, Schlachter S, Mitchell IS, Spencer KL, Banka AL, Wallace EM, et
al: Discovery and preclinical pharmacology of a selective
ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human
tumors. J Med Chem. 55:8110–8127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lin J, Sampath D, Nannini MA, Lee BB,
Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, et al:
Targeting activated Akt with GDC-0068, a novel selective Akt
inhibitor that is efficacious in multiple tumor models. Clin Cancer
Res. 19:1760–1772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kim S, Tan AR, Im S, Villanueva R, Valero
V, Saura C, Oliveira M, Isakoff SJ, Singel SM and Dent RA: LOTUS: A
randomized, phase II, multicenter, placebo-controlled study of
ipatasertib (Ipat, GDC-0068), an inhibitor of Akt, in combination
with paclitaxel (Pac) as front-line treatment for patients (pts)
with metastatic triple-negative breast cancer (TNBC). ASCO Meet
Abstr. 33:TPS11112015.
|
|
65
|
Luo Y, Shoemaker AR, Liu X, Woods KW,
Thomas SA, de Jong R, Han EK, Li T, Stoll VS, Powlas JA, et al:
Potent and selective inhibitors of Akt kinases slow the progress of
tumors in vivo. Mol Cancer Ther. 4:977–986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Woods KW, Fischer JP, Claiborne A, Li T,
Thomas SA, Zhu GD, Diebold RB, Liu X, Shi Y, Klinghofer V, et al:
Synthesis and SAR of indazole-pyridine based protein kinase B/Akt
inhibitors. Bioorg Med Chem. 14:6832–6846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin H, Yamashita DS, Xie R, Zeng J, Wang
W, Leber J, Safonov IG, Verma S, Li M, Lafrance L, et al:
Tetrasubstituted pyridines as potent and selective AKT inhibitors:
Reduced CYP450 and hERG inhibition of aminopyridines. Bioorg Med
Chem Lett. 20:684–688. 2010. View Article : Google Scholar
|
|
68
|
Ko JH, Yeon SW, Ryu JS, Kim TY, Song EH,
You HJ, Park RE and Ryu CK: Synthesis and biological evaluation of
5-arylamino-6-chloro-1H-indazole-4,7-diones as inhibitors of
protein kinase B/Akt. Bioorg Med Chem Lett. 16:6001–6005. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bencsik JR, Xiao D, Blake JF, Kallan NC,
Mitchell IS, Spencer KL, Xu R, Gloor SL, Martinson M, Risom T, et
al: Discovery of dihydrothieno- and dihydrofuropyrimidines as
potent pan Akt inhibitors. Bioorg Med Chem Lett. 20:7037–7041.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Saxty G, Woodhead SJ, Berdini V, Davies
TG, Verdonk ML, Wyatt PG, Boyle RG, Barford D, Downham R, Garrett
MD, et al: Identification of inhibitors of protein kinase B using
fragment-based lead discovery. J Med Chem. 50:2293–2296. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Grimshaw KM, Hunter L-JK, Yap TA, Heaton
SP, Walton MI, Woodhead SJ, Fazal L, Reule M, Davies TG, Seavers
LC, et al: AT7867 is a potent and oral inhibitor of AKT and p70 S6
kinase that induces pharmacodynamic changes and inhibits human
tumor xenograft growth. Mol Cancer Ther. 9:1100–1110. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yap TA, Walton MI, Grimshaw KM, Te Poele
RH, Eve PD, Valenti MR, de Haven Brandon AK, Martins V, Zetterlund
A, Heaton SP, et al: AT13148 is a novel, oral multi-AGC kinase
inhibitor with potent pharmacodynamic and antitumor activity. Clin
Cancer Res. 18:3912–3923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lin X, Murray JM, Rico AC, Wang MX, Chu
DT, Zhou Y, Del Rosario M, Kaufman S, Ma S, Fang E, et al:
Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for
AKT: Synthesis and SAR studies. Bioorg Med Chem Lett. 16:4163–4168.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dumble M, Crouthamel MC, Zhang SY, Schaber
M, Levy D, Robell K, Liu Q, Figueroa DJ, Minthorn EA, Seefeld MA,
et al: Discovery of novel AKT inhibitors with enhanced anti-tumor
effects in combination with the MEK inhibitor. PLoS One.
9:e1008802014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chang S, Zhang Z, Zhuang X, Luo J, Cao X,
Li H, Tu Z, Lu X, Ren X and Ding K: New thiazole carboxamides as
potent inhibitors of Akt kinases. Bioorg Med Chem Lett.
22:1208–1212. 2012. View Article : Google Scholar
|
|
76
|
Deng R, Yang F, Chang SH, Tang J, Qin J,
Feng GK, Ding K and Zhu XF: DC120, a novel and potent inhibitor of
AKT kinase, induces tumor cell apoptosis and suppresses tumor
growth. Mol Pharmacol. 82:189–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Spencer A, Yoon SS, Harrison SJ, Morris
SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB and Chen
C: The novel AKT inhibitor afuresertib shows favorable safety,
pharmacokinetics, and clinical activity in multiple myeloma. Blood.
124:2190–2195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Faiman B and Richards T: Innovative agents
in multiple myeloma. J Adv Pract Oncol. 5:193–202. 2014.PubMed/NCBI
|
|
79
|
Wu WI, Voegtli WC, Sturgis HL, Dizon FP,
Vigers GPA and Brandhuber BJ: Crystal structure of human AKT1 with
an allosteric inhibitor reveals a new mode of kinase inhibition.
PLoS One. 5:e129132010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lu S, Li S and Zhang J: Harnessing
allostery: A novel approach to drug discovery. Med Res Rev.
34:1242–1285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lindsley CW, Zhao Z, Leister WH, Robinson
RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber
HE, et al: Allosteric Akt (PKB) inhibitors: Discovery and SAR of
isozyme selective inhibitors. Bioorg Med Chem Lett. 15:761–764.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bilodeau MT, Balitza AE, Hoffman JM,
Manley PJ, Barnett SF, Defeo-Jones D, Haskell K, Jones RE, Leander
K, Robinson RG, et al: Allosteric inhibitors of Akt1 and Akt2: A
naphthyridinone with efficacy in an A2780 tumor xenograft model.
Bioorg Med Chem Lett. 18:3178–3182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li Y, Liang J, Siu T, Hu E, Rossi MA,
Barnett SF, Defeo-Jones D, Jones RE, Robinson RG, Leander K, et al:
Allosteric inhibitors of Akt1 and Akt2: Discovery of
[1,2,4]triazolo[3,4-f][1,6]naphthyridines with potent and balanced
activity. Bioorg Med Chem Lett. 19:834–836. 2009. View Article : Google Scholar
|
|
84
|
Hirai H, Sootome H, Nakatsuru Y, Miyama K,
Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al:
MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy
by standard chemotherapeutic agents or molecular targeted drugs in
vitro and in vivo. Mol Cancer Ther. 9:1956–1967. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Fang Z, Simard JR, Plenker D, Nguyen HD,
Phan T, Wolle P, Baumeister S and Rauh D: Discovery of inter-domain
stabilizers-a novel assay system for allosteric akt inhibitors. ACS
Chem Biol. 10:279–288. 2015. View Article : Google Scholar
|
|
86
|
Jiao P, Zhou YS, Yang JX, Zhao YL, Liu QQ,
Yuan C and Wang FZ: MK-2206 induces cell cycle arrest and apoptosis
in HepG2 cells and sensitizes TRAIL-mediated cell death. Mol Cell
Biochem. 382:217–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Molife LR, Yan L, Vitfell-Rasmussen J,
Zernhelt AM, Sullivan DM, Cassier PA, Chen E, Biondo A, Tetteh E,
Siu LL, et al: Phase 1 trial of the oral AKT inhibitor MK-2206 plus
carboplatin/paclitaxel, docetaxel, or erlotinib in patients with
advanced solid tumors. J Hematol Oncol. 7:12014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kümler I, Tuxen MK and Nielsen DL: A
systematic review of dual targeting in HER2-positive breast cancer.
Cancer Treat Rev. 40:259–270. 2014. View Article : Google Scholar
|
|
89
|
Konopleva MY, Walter RB, Faderl SH,
Jabbour EJ, Zeng Z, Borthakur G, Huang X, Kadia TM, Ruvolo PP,
Feliu JB, et al: Preclinical and early clinical evaluation of the
oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous
leukemia. Clin Cancer Res. 20:2226–2235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Do K, Speranza G, Bishop R, Khin S,
Rubinstein L, Kinders RJ, Datiles M, Eugeni M, Lam MH, Doyle LA, et
al: Biomarker-driven phase 2 study of MK-2206 and selumetinib
(AZD6244, ARRY-142886) in patients with colorectal cancer. Invest
New Drugs. 33:720–728. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Meuillet EJ: Novel inhibitors of AKT:
Assessment of a different approach targeting the pleckstrin
homology domain. Curr Med Chem. 18:2727–2742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gills JJ and Dennis PA: Perifosine: Update
on a novel Akt inhibitor. Curr Oncol Rep. 11:102–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
van Blitterswijk WJ and Verheij M:
Anticancer mechanisms and clinical application of
alkylphospholipids. Biochim Biophys Acta. 1831.663–674. 2013.
|
|
94
|
van der Luit AH, Vink SR, Klarenbeek JB,
Perrissoud D, Solary E, Verheij M and van Blitterswijk WJ: A new
class of anticancer alkylphospholipids uses lipid rafts as membrane
gateways to induce apoptosis in lymphoma cells. Mol Cancer Ther.
6:2337–2345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Reis-Sobreiro M, Roué G, Moros A, Gajate
C, de la Iglesia-Vicente J, Colomer D and Mollinedo F: Lipid
raft-mediated Akt signaling as a therapeutic target in mantle cell
lymphoma. Blood Cancer J. 3:e1182013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
de Pachioni JA, Magalhães JG, Lima EJC, de
Bueno LM, Barbosa JF, de Sá MM and Rangel-Yagui CO:
Alkylphospholipids - a promising class of chemotherapeutic agents
with a broad pharmacological spectrum. J Pharm Pharm Sci.
16:742–759. 2013.
|
|
97
|
Giantonio BJ, Derry C, McAleer C,
McPhillips JJ and O‘Dwyer PJ: Phase I and pharmacokinetic study of
the cytotoxic ether lipid ilmofosine administered by weekly
two-hour infusion in patients with advanced solid tumors. Clin
Cancer Res. 10:1282–1288. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Verweij J, Krzemieniecki K, Kok T, Poveda
A, van Pottelsberghe C, van Glabbeke M and Mouridsen H: Phase II
study of miltefosine (hexadecylphosphocholine) in advanced soft
tissue sarcomas of the adult - an EORTC Soft Tissue and Bone
Sarcoma Group Study. Eur J Cancer. 29A:208–209. 1993. View Article : Google Scholar
|
|
99
|
Becher R, Kloke K, Füger A, Bremer K,
Drozd A, Kleeberg UR, Fritze D, Rieche K and Sindermann H: Phase II
Trial of Orally Administered Miltefosine in Advanced Colorectal
Cancer. Onkologie. 16:11–15. 1993. View Article : Google Scholar
|
|
100
|
Clive S, Gardiner J and Leonard RCF:
Miltefosine as a topical treatment for cutaneous metastases in
breast carcinoma. Cancer Chemother Pharmacol Suppl. 44:2000.
|
|
101
|
Dorlo TPC, Balasegaram M, Beijnen JH and
de Vries PJ: Miltefosine: A review of its pharmacology and
therapeutic efficacy in the treatment of leishmaniasis. J
Antimicrob Chemother. 67:2576–2597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Orlowski RZ: Novel agents for multiple
myeloma to overcome resistance in phase III clinical trials. Semin
Oncol. 40:634–651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Henke G, Lindner LH, Vogeser M, Eibl HJ,
Wörner J, Müller AC, Bamberg M, Wachholz K, Belka C and Jendrossek
V: Pharmacokinetics and biodistribution of Erufosine in nude mice -
implications for combination with radiotherapy. Radiat Oncol.
4:462009. View Article : Google Scholar :
|
|
104
|
Rübel A, Handrick R, Lindner LH, Steiger
M, Eibl H, Budach W, Belka C and Jendrossek V: The membrane
targeted apoptosis modulators erucylphosphocholine and
erucylphosphohomocholine increase the radiation response of human
glioblastoma cell lines in vitro. Radiat Oncol. 1:62006. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kaleağasıoğlu F and Berger MR:
Differential effects of erufosine on proliferation, wound healing
and apoptosis in colorectal cancer cell lines. Oncol Rep.
31:1407–1416. 2014.
|
|
106
|
Rudner J, Ruiner CE, Handrick R, Eibl HJ,
Belka C and Jendrossek V: The Akt-inhibitor Erufosine induces
apoptotic cell death in prostate cancer cells and increases the
short term effects of ionizing radiation. Radiat Oncol. 5:1082010.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chinni SR and Sarkar FH: Akt inactivation
is a key event in indole-3-carbinol-induced apoptosis in PC-3
cells. Clin Cancer Res. 8:1228–1236. 2002.PubMed/NCBI
|
|
108
|
Aggarwal BB and Ichikawa H: Molecular
targets and anticancer potential of indole-3-carbinol and its
derivatives. Cell Cycle. 4:1201–1215. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Kim DJ, Reddy K, Kim MO, Li Y, Nadas J,
Cho YY, Kim JE, Shim JH, Song NR, Carper A, et al:
(3-Chloroacetyl)-indole, a novel allosteric AKT inhibitor,
suppresses colon cancer growth in vitro and in vivo. Cancer Prev
Res (Phila). 4:1842–1851. 2011. View Article : Google Scholar
|
|
110
|
Chao WR, Yean D, Amin K, Green C and Jong
L: Computer-aided rational drug design: A novel agent (SR13668)
designed to mimic the unique anticancer mechanisms of dietary
indole-3-carbinol to block Akt signaling. J Med Chem. 50:3412–3415.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kapetanovic IM, Muzzio M, Hu S-C, Crowell
JA, Rajewski RA, Haslam JL, Jong L and McCormick DL:
Pharmacokinetics and enhanced bioavailability of candidate cancer
preventative agent, SR13668 in dogs and monkeys. Cancer Chemother
Pharmacol. 65:1109–1116. 2010. View Article : Google Scholar
|
|
112
|
Reid JM, Walden CA, Qin R, Ziegler KL,
Haslam JL, Rajewski RA, Warndahl R, Fitting CL, Boring D, Szabo E,
et al; Cancer Prevention Network. Phase 0 clinical chemoprevention
trial of the Akt inhibitor SR13668. Cancer Prev Res (Phila).
4:347–353. 2011. View Article : Google Scholar
|
|
113
|
Weng JR, Tsai CH, Omar HA, Sargeant AM,
Wang D, Kulp SK, Shapiro CL and Chen CS: OSU-A9, a potent
indole-3-carbinol derivative, suppresses breast tumor growth by
targeting the Akt-NF-kappaB pathway and stress response signaling.
Carcinogenesis. 30:1702–1709. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mahadevan D, Powis G, Mash EA, George B,
Gokhale VM, Zhang S, Shakalya K, Du-Cuny L, Berggren M, Ali MA, et
al: Discovery of a novel class of AKT pleckstrin homology domain
inhibitors. Mol Cancer Ther. 7:2621–2632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Moses SA, Ali MA, Zuohe S, Du-Cuny L, Zhou
LL, Lemos R, Ihle N, Skillman AG, Zhang S, Mash EA, et al: In vitro
and in vivo activity of novel small-molecule inhibitors targeting
the pleckstrin homology domain of protein kinase B/AKT. Cancer Res.
69:5073–5081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Meuillet EJ, Zuohe S, Lemos R, Ihle N,
Kingston J, Watkins R, Moses SA, Zhang S, Du-Cuny L and Herbst R:
Molecular pharmacology and antitumor activity of PHT-427, a novel
Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin
homology domain inhibitor. Mol Cancer Ther. 9:706–717. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Miao B, Skidan I, Yang J, Lugovskoy A,
Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, et
al: Small molecule inhibition of
phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to
pleckstrin homology domains. Proc Natl Acad Sci USA.
107:20126–20131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kommagalla Y, Cornea S, Riehle R,
Torchilin V, Degterev A and Ramana CV: Optimization of the
anti-cancer activity of phosphatidylinositol-3 kinase pathway
inhibitor PITENIN-1: Switching a thiourea with 1,2,3-triazole.
MedChemComm. 5:1359–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nitulescu GM, Draghici C, Olaru OT, Matei
L, Ioana A, Dragu LD and Bleotu C: Synthesis and apoptotic activity
of new pyrazole derivatives in cancer cell lines. Bioorg Med Chem.
23:5799–5808. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang L, Dan HC, Sun M, Liu Q, Sun XM,
Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, et al:
Akt/protein kinase B signaling inhibitor-2, a selective small
molecule inhibitor of Akt signaling with antitumor activity in
cancer cells overexpressing Akt. Cancer Res. 64:4394–4399. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Sampath D, Malik A, Plunkett W, Nowak B,
Williams B, Burton M, Verstovsek S, Faderl S, Garcia-Manero G, List
AF, et al: Phase I clinical, pharmacokinetic, and pharmacodynamic
study of the Akt-inhibitor triciribine phosphate monohydrate in
patients with advanced hematologic malignancies. Leuk Res.
37:1461–1467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Berndt N, Yang H, Trinczek B, Betzi S,
Zhang Z, Wu B, Lawrence NJ, Pellecchia M, Schönbrunn E, Cheng JQ,
et al: The Akt activation inhibitor TCN-P inhibits Akt
phosphorylation by binding to the PH domain of Akt and blocking its
recruitment to the plasma membrane. Cell Death Differ.
17:1795–1804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Evangelisti C, Ricci F, Tazzari P,
Chiarini F, Battistelli M, Falcieri E, Ognibene A, Pagliaro P,
Cocco L, McCubrey JA, et al: Preclinical testing of the Akt
inhibitor triciribine in T-cell acute lymphoblastic leukemia. J
Cell Physiol. 226:822–831. 2011. View Article : Google Scholar
|
|
124
|
Dieterle A, Orth R, Daubrawa M, Grotemeier
A, Alers S, Ullrich S, Lammers R, Wesselborg S and Stork B: The Akt
inhibitor triciribine sensitizes prostate carcinoma cells to
TRAIL-induced apoptosis. Int J Cancer. 125:932–941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kim R, Yamauchi T, Husain K, Sebti S and
Malafa M: Triciribine Phosphate Monohydrate, an AKT Inhibitor,
Enhances Gemcitabine Activity in Pancreatic Cancer Cells.
Anticancer Res. 35:4599–4604. 2015.PubMed/NCBI
|
|
126
|
Kim D, Sun M, He L, Zhou QH, Chen J, Sun
XM, Bepler G, Sebti SM and Cheng JQ: A small molecule inhibits Akt
through direct binding to Akt and preventing Akt membrane
translocation. J Biol Chem. 285:8383–8394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Li B, Ren H, Yue P, Chen M, Khuri FR and
Sun SY: The novel Akt inhibitor API-1 induces c-FLIP degradation
and synergizes with TRAIL to augment apoptosis independent of Akt
inhibition. Cancer Prev Res (Phila). 5:612–620. 2012. View Article : Google Scholar
|
|
128
|
Ashwell MA, Lapierre J-M, Brassard C,
Bresciano K, Bull C, Cornell-Kennon S, Eathiraj S, France DS, Hall
T, Hill J, et al: Discovery and optimization of a series of
3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: Orally
bioavailable, selective, and potent ATP-independent Akt inhibitors.
J Med Chem. 55:5291–5310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Chan TCK, Lapierre JM, Ashwell MA, France
DS, Chen CR, Cornell-Kennon S, Bull C, Eathiraj S, Palma R, Liu Y,
et al: Abstract A230: Discovery and characterization of ARQ 092, an
ATP-independent, potent and selective inhibitor of AKT kinases. Mol
Cancer Ther. 10(Suppl 1): A230. 2011. View Article : Google Scholar
|
|
130
|
Yu Y, Savage RE, Eathiraj S, Meade J, Wick
MJ, Hall T, Abbadessa G and Schwartz B: Targeting AKT1-E17K and the
PI3K/AKT pathway with an allosteric AKT inhibitor, ARQ 092. PLoS
One. 10:e01404792015. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Politz O, Scholz A, Haegebarth A, Liu N,
Baerfacker L, Ince S, Neuhaus R, Boemer U, Michels M and Mumberg D:
Abstract 3685: BAY 1125976, is a selective allosteric AKT1/2
inhibitor with high efficacy in AKT1-mutated cancers. Cancer Res.
74(Suppl 19): 36852014. View Article : Google Scholar
|
|
132
|
Yilmaz OG, Olmez EO and Ulgen KO:
Targeting the Akt1 allosteric site to identify novel scaffolds
through virtual screening. Comput Biol Chem. 48:1–13. 2014.
View Article : Google Scholar
|
|
133
|
Estrada AC, Syrovets T, Pitterle K, Lunov
O, Büchele B, Schimana-Pfeifer J, Schmidt T, Morad SA and Simmet T:
Tirucallic acids are novel pleckstrin homology domain-dependent Akt
inhibitors inducing apoptosis in prostate cancer cells. Mol
Pharmacol. 77:378–387. 2010. View Article : Google Scholar
|
|
134
|
Morrow JK, Du-Cuny L, Chen L, Meuillet EJ,
Mash EA, Powis G and Zhang S: Recent development of anticancer
therapeutics targeting Akt. Recent Patents Anticancer Drug Discov.
6:146–159. 2011. View Article : Google Scholar
|
|
135
|
Toral-Barza L, Zhang WG, Huang X, McDonald
LA, Salaski EJ, Barbieri LR, Ding WD, Krishnamurthy G, Hu YB, Lucas
J, et al: Discovery of lactoquinomycin and related
pyranonaphthoquinones as potent and allosteric inhibitors of
AKT/PKB: Mechanistic involvement of AKT catalytic activation loop
cysteines. Mol Cancer Ther. 6:3028–3038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Salaski EJ, Krishnamurthy G, Ding WD, Yu
K, Insaf SS, Eid C, Shim J, Levin JI, Tabei K, Toral-Barza L, et
al: Pyranonaphthoquinone lactones: A new class of AKT selective
kinase inhibitors alkylate a regulatory loop cysteine. J Med Chem.
52:2181–2184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Nguyen T, Coover RA, Verghese J, Moran RG
and Ellis KC: Phenylalanine-Based Inactivator of AKT Kinase:
Design, synthesis, and biological evaluation. ACS Med Chem Lett.
5:462–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Shearn CT, Reigan P and Petersen DR:
Inhibition of hydrogen peroxide signaling by 4-hydroxynonenal due
to differential regulation of Akt1 and Akt2 contributes to
decreases in cell survival and proliferation in hepatocellular
carcinoma cells. Free Radic Biol Med. 53:1–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Weisner J, Gontla R, van der Westhuizen L,
Oeck S, Ketzer J, Janning P, Richters A, Mühlenberg T, Fang Z,
Taher A, et al: Covalent-allosteric kinase inhibitors. Angew Chem
Int Ed Engl. 54:10313–10316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
She QB, Chandarlapaty S, Ye Q, Lobo J,
Haskell KM, Leander KR, DeFeo-Jones D, Huber HE and Rosen N: Breast
tumor cells with PI3K mutation or HER2 amplification are
selectively addicted to Akt signaling. PLoS One. 3:e30652008.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Sangai T, Akcakanat A, Chen H, Tarco E, Wu
Y, Do KA, Miller TW, Arteaga CL, Mills GB, Gonzalez-Angulo AM and
Meric-Bernstam F: Biomarkers of response to Akt inhibitor MK-2206
in breast cancer. Clin Cancer Res. 18:5816–5828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Davies BR, Greenwood H, Dudley P, Crafter
C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, et al: Preclinical
pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics,
antitumor activity, and correlation of monotherapy activity with
genetic background. Mol Cancer Ther. 11:873–887. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Yap TA, Yan L, Patnaik A, Fearen I, Olmos
D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, et
al: First-in-man clinical trial of the oral pan-AKT inhibitor
MK-2206 in patients with advanced solid tumors. J Clin Oncol.
29:4688–4695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Saura C, Jones S, Mateo J, Hollebecque A,
Cleary JM, Perez DR, Zhu J, Musib LC, Patel PH, Cervantes-Ruiperez
A, et al: A phase Ib study of the Akt inhibitor GDC-0068 with
docetaxel (D) or mFOLFOX-6 (F) in patients (pts) with advanced
solid tumors. J Clin Oncol. 30:Suppl; abstr 3021. 2012.
|
|
145
|
Yan Y, Wagle M, Punnoose E, Musib L, Budha
N, Nannini M, Lin K, Liederer BM, Murli S, Ramakrishnan V, et al: A
first-inhuman trial of GDC-0068: A novel, oral, ATP-competitive Akt
inhibitor, demonstrates robust suppression of the Akt pathway in
surrogate and tumor tissues. Mol Cancer Ther. 10:abstr B154. 2011.
View Article : Google Scholar
|
|
146
|
Banerji U, Ranson M, Schellens J,
Schellens JH, Esaki T, Dean EJ and Zivi A: Results of two phase I
multicenter trials of AZD5363, an inhibitor of AKT1, 2 and 3:
Biomarker and early clinical evaluation in Western and Japanese
patients with advanced solid tumors. Cancer Res. 73:abstr LB-66.
2013. View Article : Google Scholar
|
|
147
|
Dienstmann R, Rodon J, Serra V and
Tabernero J: Picking the point of inhibition: A comparative review
of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 13:1021–1031.
2014. View Article : Google Scholar : PubMed/NCBI
|