Introduction
Erlotinib belongs to the class of molecular targeted
drugs designed as epidermal growth factor receptor (EGFR) tyrosine
kinase inhibitors (TKIs). It blocks trans-phosphorylation of EGFR
and subsequent downstream signaling in pathways such as the
mitogen-activated protein kinase (MAPK) pathway,
phosphatidylinositol 3-kinase (PI3K)-AKT pathway, and signal
transducer and activator of transcription 3 (STAT3) pathway.
Erlotinib treatment results in prolonged progression-free survival
(PFS) with a median of 10–14 months in patients with non-small cell
lung cancer (NSCLC) harboring EGFR exon 19 deletion or L858R
mutations (EGFR Mut+ NSCLC) (1–3).
However, despite these clinical benefits, almost all tumors
eventually progress due to acquired resistance (4). Recently, several mechanisms of
EGFR-TKI resistance have been identified, including EGFR
T790M gatekeeper mutation, activation of bypass signals
(ERBB2 gene amplification and MET gene
amplification), and other mechanisms (transformation to small cell
lung cancer, epithelial to mesenchymal transition, and tumor
microenvironment-mediated resistance) (5).
Bevacizumab, a humanized monoclonal antibody
targeting vascular endothelial growth factor (VEGF), regresses
preexisting tumor blood vessels and blocks the formation of new
ones (6,7). Furthermore, it normalizes vascular
permeability and thereby decreases interstitial fluid pressure so
that it improves delivery of co-administered drugs and therapeutic
outcomes (8–10). Consequently, bevacizumab prolongs
PFS and overall survival in advanced NSCLC when administered in
combination with standard first-line platinum-based chemo-therapies
(11).
Since erlotinib and bevacizumab act on two different
pathways critical to tumor growth, administering these drugs
concomitantly may confer promising clinical benefits to cancer
patients with advanced disease (12,13).
The Phase II JO25567 study reported that erlotinib plus bevacizumab
produced a statistically significant and clinically meaningful
prolongation of PFS compared with erlotinib alone in the treatment
of EGFR Mut+ NSCLC (14). Several preclinical studies in
various xenograft models have reported on the mechanisms of
erlotinib in addition to bevacizumab (15). In those studies, erlotinib was
shown to decrease VEGF expression (16,17)
and block synthesis of angiogenic proteins such as basic fibroblast
growth factor (bFGF) and transforming growth factor-α (TGF-α)
(12,18). Moreover, PTK787, an inhibitor of
VEGF receptor (VEGFR) tyrosine kinases, c-Kit, and angiogenesis,
was shown to improve delivery of erlotinib into the tumor in a PC-9
xenograft model (19). However,
those data show the mechanisms in the erlotinib-sensitive phase,
and the mechanism by which the combination of erlotinib and
bevacizumab confers prolonged efficacy even into the
erlotinib-refractory phase remains to be elucidated.
In the present study, we established a human
EGFR Mut+ NSCLC xenograft model that became
refractory in which tumor regrowth was observed by long-term
erlotinib administration, and we analyzed the mechanisms of both
the erlotinib-sensitive and erlotinib-refractory phases.
Materials and methods
Test agents
Erlotinib was provided by F. Hoffmann-La Roche Ltd.
(Basel, Switzerland) and was dissolved in 6% Captisol solution
(ChemScene, Monmouth Junction, NJ, USA). Bevacizumab was obtained
from F. Hoffmann-La Roche Ltd. Human immunoglobulin G (HuIgG) was
purchased from MP Biomedicals (Santa Ana, CA, USA). Both
bevacizumab and HuIgG were diluted with saline.
Cell lines and culture conditions
B901L (harboring EGFR exon 19 deletion) was
purchased from the institute of Physical and Chemical Research
(RIKEN, Saitama, Japan). This cell line was maintained in RPMI-1640
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v)
fetal bovine serum (Bovogen Biologicals, Melbourne, Australia),
0.45% D-glucose (Sigma-Aldrich), 10 mM HEPES buffer
(Sigma-Aldrich), and 1 mM Na-pyruvate (Thermo Fisher Scientific,
Waltham, MA, USA) at 37°C under 5% CO2. NCI-H1975
(harboring T790M mutation) was purchased from ATCC and maintained
in RPMI-1640 supplemented with 10% (v/v) fetal bovine serum at 37°C
under 5% CO2.
Animals
Male, 5-week-old BALB/c-nu/nu mice (CAnN.
Cg-Foxn1<nu>/CrlCrlj nu/nu) were obtained from Charles River
Laboratories Inc. (Kanagawa, Japan). All animals were allowed to
acclimatize and recover from shipping-related stress for at least 1
week prior to the study. The health of the mice was monitored by
daily observation. The animals were kept under a controlled
light-dark cycle (12–12 h), and chlorinated water and irradiated
food were provided ad libitum. All animal experiments were
reviewed and approved by the Institutional Animal Care and Use
Committee at Chugai Pharmaceutical Co., Ltd.
Evaluation of antitumor activity with
concurrent treatment
Each mouse was subcutaneously inoculated into the
right flank with B901L cells (5×106 cells/mouse). After
the tumor volume (TV) reached 200–600 mm3, mice were
randomly allocated to control and treatment groups, and
administration of drugs was started (day 1). Bevacizumab or HuIgG
was administered intraperitoneally once a week at a maximum
effective dose of 5 mg/kg. Erlotinib or vehicle was orally
administered daily at 30 or 60 mg/kg (maximum effective dose). TV
and body weight were measured twice a week. The antitumor activity
was evaluated by TV, which was estimated from the equation TV =
ab2/2, where a and b are tumor length and width,
respectively. Tumor regrowth was defined as an increase in TV at
the final observation date compared to the day of minimum TV.
Complete tumor regression was defined as a tumor volume below the
limit of detection of <15 mm3.
Evaluation of antitumor activity with
bevacizumab add-on treatment after becoming
erlotinib-refractory
B901L cells (5×106 cells/mouse) were
inoculated subcutaneously into the right flank of the mice. After
TV reached 200–600 mm3, mice were administered erlotinib
orally daily from day 1 until day 63. The individual mice which
tumor regrowth was observed was re-randomized on day 64 and
allocated to erlotinib, bevacizumab, or combination of erlotinib
plus bevacizumab groups. Erlotinib or vehicle was administered
orally daily from day 64 to day 78. Bevacizumab or HuIgG was
administered intraperitoneally once a week from day 64 to day 78.
To evaluate the antitumor effect, TV and body weight were measured
twice a week. The antitumor activity was evaluated by TV ratio,
which was calculated by the equation a/b, where a is the TV each
day and b is the TV on day 64 because the TV of the largest tumor
in the re-randomized group on day 64 was more than twice that of
the smallest tumor. The relative TV mean of day 22 became minimum.
Complete tumor regression was defined as tumor volume under
detection limit <15 mm3.
Tumor tissue homogenates
Tumor tissues were collected 3 h after erlotinib
administration on indicated days and were immediately frozen in
liquid nitrogen and stored at −80°C until use. The tumor tissues
were homogenized with Cell Lysis Buffer (Cell Signaling Technology,
Danvers, MA, USA) containing NaF (Sigma-Aldrich), Complete Protease
Inhibitor Cocktail Tablets (Roche Diagnostics, Tokyo, Japan), and
PhosSTOP (Roche Diagnostics). Following centrifugation, the
resultant supernatant was used for the assays. Total protein
concentration of the supernatant was quantified with a Direct
Detect spectrometer (Merck Millipore, Darmstadt, Germany).
Western blotting
Tumor tissue homogenate supernatants (20 μg
protein/lane) were electrophoresed on SDS-PAGE and transferred to a
polyvinylidene difluoride membrane using an iBlot Gel Transfer
Device (Thermo Fisher Scientific). The membrane was blocked in
Blocking One (Nacalai Tesque, Kyoto, Japan) and was primarily
treated with antibodies to EGFR, STAT3, AKT, ERK, pEGFR, pAKT, and
pERK (Cell Signaling Technology), antibody to pSTAT3 (Abcam,
Cambridge, MA, USA), and antibody to β-actin (Sigma-Aldrich). These
proteins were detected by horseradish peroxidase-conjugated
secondary antibodies (Cell Signaling Technology) and ECL Prime
Western Blotting Detection Reagents (GE Healthcare Life Sciences,
Little Chalfont, UK). ImageQuant 400 (GE Healthcare Life Sciences)
was used for detection, and ImageQuant TL Software was used to
digitize the strength of bands.
ELISA analysis
The concentrations of human VEGF, bFGF, placental
growth factor (PlGF), TGF-α, granulocyte-colony stimulating factor
(G-CSF), and interleukin-6 (IL-6) in homogenates of tumor tissues
were evaluated by using Quantikine ELISA (R&D Systems,
Minneapolis, MN, USA) following the manufacturer's instructions.
The human chemokine (C-X-C motif) ligand 2 (CXCL2) concentrations
in tumor tissues were evaluated by ELISA kit (Abnova, Taipei,
Taiwan) following the manufacturer's instructions. A Benchmark Plus
Microplate Reader (Bio-Rad, Tokyo, Japan) was used for
detection.
Ki-67 staining
Proliferating cells were assessed with
immunohistochemical staining of Ki-67 (mouse anti-human Ki-67
monoclonal antibody; Agilent Technologies, Glostrup, Denmark).
B901L tumors were collected on day 6 after initiation of the
treatment. The tissues were fixed with 10% neutral buffered
formalin, and embedded in paraffin. Ki-67 staining was performed
and the number of Ki-67+ tumor cells in 1000 tumor cells
was counted by Sapporo General Pathology Laboratory Co., Ltd.
(Sapporo, Japan).
Quantification of microvessel density in
tumor tissues
Microvessel density (MVD) in tumor tissues was
evaluated by immunohistochemical staining of CD31 (rat anti-mouse
CD31 monoclonal antibody; BD Biosciences, San Jose, CA, USA). Tumor
samples from freshly frozen tissues were collected on indicated
days. MVD (%) was calculated from the ratio of the CD31-positive
staining area to the total observation area in the viable region.
Three to six fields per section were randomly analyzed, excluding
necrotic areas. Positive staining areas were calculated by using
imaging analysis software (WinROOF; Mitani Corporation, Fukui,
Japan).
Melting curve analysis
Melting curve analysis was performed as previously
described (20). In brief, to
analyze T790M mutation status, exon 20 of EGFR was first
amplified by PCR from DNA by using the appropriate primers and the
LightCycler 480 Genotyping Master (Roche Diagnostics, Mannheim,
Germany), and then hybridized by using sensor and anchor probes.
Human genomic DNA (Promega, Madison, WI, USA) was used as a
wild-type EGFR control.
Statistical analysis
Differences were considered statistically
significant for values of P<0.05 by the t-test or Wilcoxon's
rank sum test using JMP version 11 software (SAS Institute, Cary,
NC, USA). Data are represented as means and SD.
Results
Combined administration of erlotinib plus
bevacizumab resulted in prolonged antitumor efficacy in B901L
xenograft model
First, we evaluated the dose response of erlotinib
in the B901L xenograft model. Although significant tumor growth
inhibition was observed in mice treated with 30 mg/kg, drug
efficacy was moderate and did not result in significant tumor
shrinkage (data not shown). In mice treated with 60 mg/kg,
on the other hand, remarkable tumor shrinkage was observed in the
initial phase. However, tumor regrowth was observed despite
continued erlotinib administration (Fig. 1). It is known that almost all
patients eventually acquire resistance to EGFR-TKIs within a few
years (4), and with this B901L
xenograft model we successfully established a model that could
replicate this acquired resistance to erlotinib by prolonged
treatment.
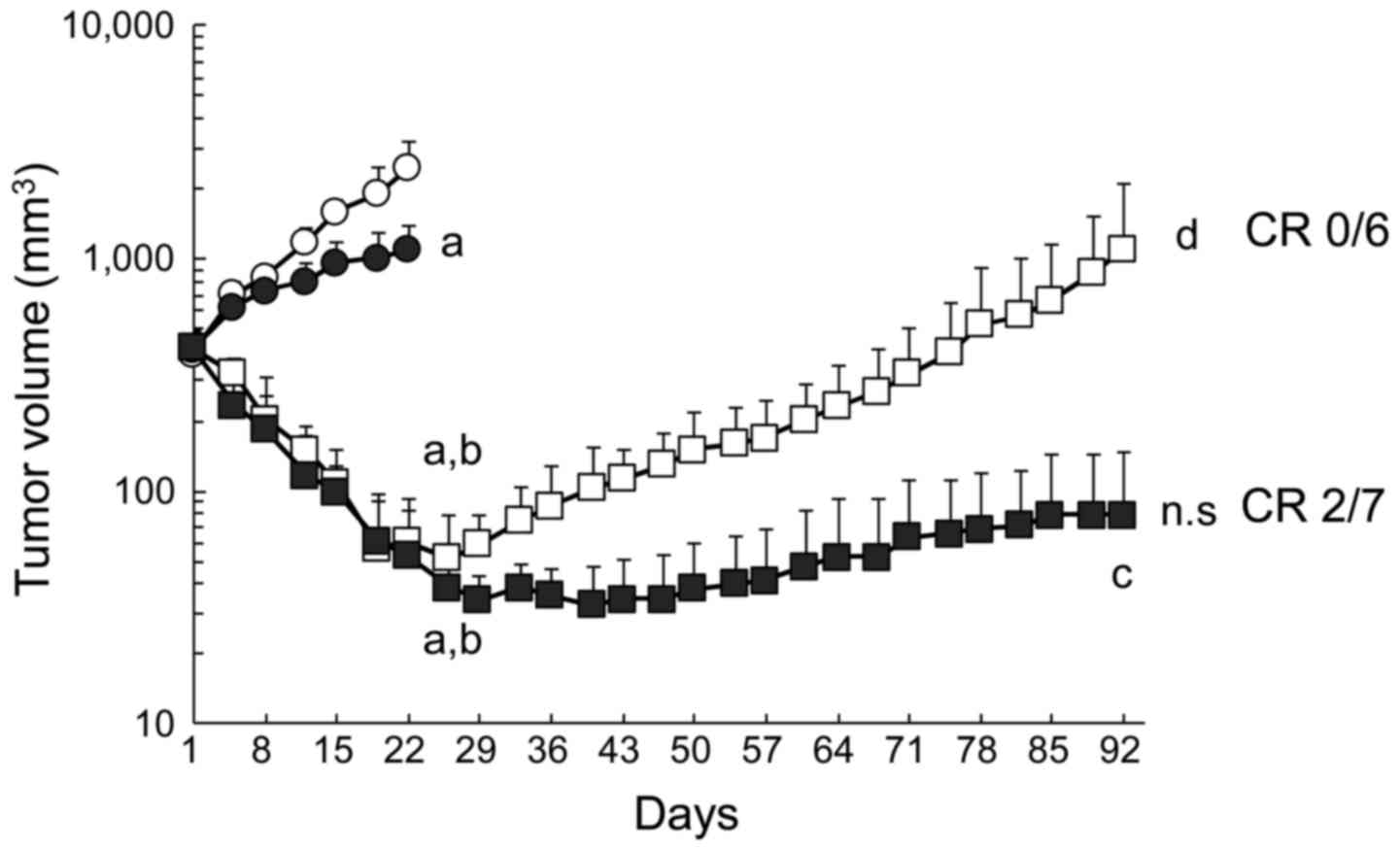 | Figure 1Prolonged antitumor efficacy of
erlotinib plus bevacizumab. B901L xenograft-bearing mice were
continuously treated with control (HuigG plus vehicle, open
circles), 5 mg/kg of bevacizumab (closed circles), 60 mg/kg of
erlotinib (open squares), or erlotinib plus bevacizumab
(combination, closed squares) (n=6 or 7). Each point represents the
mean + SD. a, P<0.05 versus control on day 22; b, P<0.05
versus bevacizumab on day 22; c, P<0.05 versus erlotinib on day
92 (by Wilcoxon test). d, P<0.05 versus erlotinib on day 26;
n.s., P>0.05 versus combination on day 40 when the mean TV of
each group reached its respective minimum (by Wilcoxon test). CR,
complete tumor regression. |
Next, we investigated the effects of combined
administration of bevacizumab in this model. In distinction to
erlotinib, no significant tumor regrowth was observed in the
erlotinib plus bevacizumab group at the final observation date.
Complete tumor regression, which was defined as a tumor volume
below the detection limit, was observed in 2/7 mice treated with
erlotinib plus bevacizumab, whereas complete tumor regression was
not observed in any mice treated with erlotinib alone.
Erlotinib and bevacizumab exhibit
stronger inhibition of MVD and tumor cell proliferation in the
erlotinib-sensitive phase
In the erlotinib-sensitive phase, remarkable tumor
regression was observed with both the erlotinib and the erlotinib
plus bevacizumab treatments. Even though there was no difference in
tumor volume in the erlotinib-sensitive phase, afterwards a
superior antitumor effect was observed and complete tumor
regression was observed in some mice in the erlotinib plus
bevacizumab group in contrast to the tumor regrowth seen in the
erlotinib group (Fig. 1).
Therefore, we supposed that cellular changes had already occurred
in the tumor even in the earlier phase.
First we examined the effect on signal transduction
by using tumor tissues obtained on day 5. EGFR and downstream ERK,
AKT, and STAT3 phosphorylation were inhibited in the erlotinib and
erlotinib plus bevacizumab groups. On the other hand, inhibition of
signal transduction was not detected in the bevacizumab group
(Fig. 2A).
 | Figure 2Effect of erlotinib and bevacizumab
on signal transduction and histological analyses. (A) Immunoblots
of tumor lysates 3 h after treatment with control, 5 mg/kg of
bevacizumab, 60 mg/kg of erlotinib, or erlotinib plus bevacizumab
combination. Tumor samples (n=6) were collected on day 5. Total
EGFR (tEGFR), phosphorylated EGFR (pEGFR), total ERK (tERK),
phosphorylated ERK (pERK), total AKT (tAKT), phosphorylated AKT
(pAKT), total STAT3 (tSTAT3), phosphorylated STAT3 (pSTAT3), and
actin were detected by a manual western blotting method. (B)
Representative images of tumor samples (n=12) sectioned on day 6
and stained with Ki-67. (C) Number of Ki-67+ tumor cells
per 1000 tumor cells. (D) MVD in tumor tissues was determined by
CD31-immunostaining. Representative immunohistochemical images of
tumor samples collected on day 4 (n=5 or 8). Scale bar is 100
μm. (e) MVD in the control, bevacizumab, erlotinib, and
erlotinib plus bevacizumab groups. Bars represent mean ± SD. a,
P<0.05 versus control; b, P<0.05 versus bevacizumab; c,
P<0.05 versus erlotinib (by Wilcoxon test). |
Next, we determined whether there was any difference
in tumor cell proliferation in tumor tissues sampled on day 6.
Compared with control, the number of Ki-67+ tumor cells
in the area was decreased by each single agent. With erlotinib plus
bevacizumab, it was further decreased compared with each drug alone
(Fig. 2B). The number of
Ki-67+ tumor cells per 1000 tumor cells (mean ± SD) was
also decreased significantly in the erlotinib plus bevacizumab
group (308±74) compared to that in the control (610±74),
bevacizumab (488±63), and erlotinib (426±84) groups (P<0.05)
(Fig. 2C).
Then, MVD in tumor tissues was evaluated using
specimens obtained on day 4. MVD (%; mean ± SD) of the control,
bevacizumab, erlotinib, and erlotinib plus bevacizumab groups was,
respectively, 3.79±0.58, 1.85±0.53, 2.88±0.43, and 1.35±0.28,
indicating that erlotinib plus bevacizumab suppressed MVD compared
with each drug alone (P<0.05) (Fig.
2D and E). Among the cytokines with pro-angiogenic activities
that we tested, IL-6, G-CSF, and CXCL2 were inhibited by erlotinib,
whereas bFGF and TGF-α were not (Table
I).
 | Table ILevels of angiogenic factors in
different phases of erlotinib treatment. |
Table I
Levels of angiogenic factors in
different phases of erlotinib treatment.
| Angiogenic
factor | Erlotinib-sensitive
phase
|
Erlotinib-refractory phase
|
|---|
| Control | Erlotinib | Erlotinib |
|---|
| bFGF | 194±58.6 | 189±104 | 201±78.6 |
| PlGF | N.D. | N.D. | N.D. |
| TGF-α | 34.1±7.59 | 31.1±8.32 | N.D. |
| G-CSF | 269±154 | 62.4±59.3a | 99.8±76.4 |
| IL-6 | 924±1022 | 22.6±8.94a | 80.3±51.1a,b |
| CXCL2 | 223±43.8 | 19.9±4.54a | 29.7±4.29a |
Re-induction of tumor VEGF was involved
in the erlotinib resistance mechanism and was inhibited by
bevacizumab
As shown in Fig. 1,
tumor became refractory and regrowth was observed in the erlotinib
group. Therefore, we compared the status of signal transduction
using specimens collected in the erlotinib-sensitive and
erlotinib-refractory phases. In the erlotinib-sensitive phase (day
5), pEGFR, pERK, pAKT, and pSTAT3 were strongly decreased. In
contrast, although pEGFR was still suppressed in the erlotinib
group in the erlotinib-refractory phase (day 75), pERK, pAKT, and
pSTAT3 were increased compared with levels on day 5 (Fig. 3A). This finding implied that there
was a resistance mechanism other than that occurring in EGFR
itself, although the T790M mutation was not detected in this model
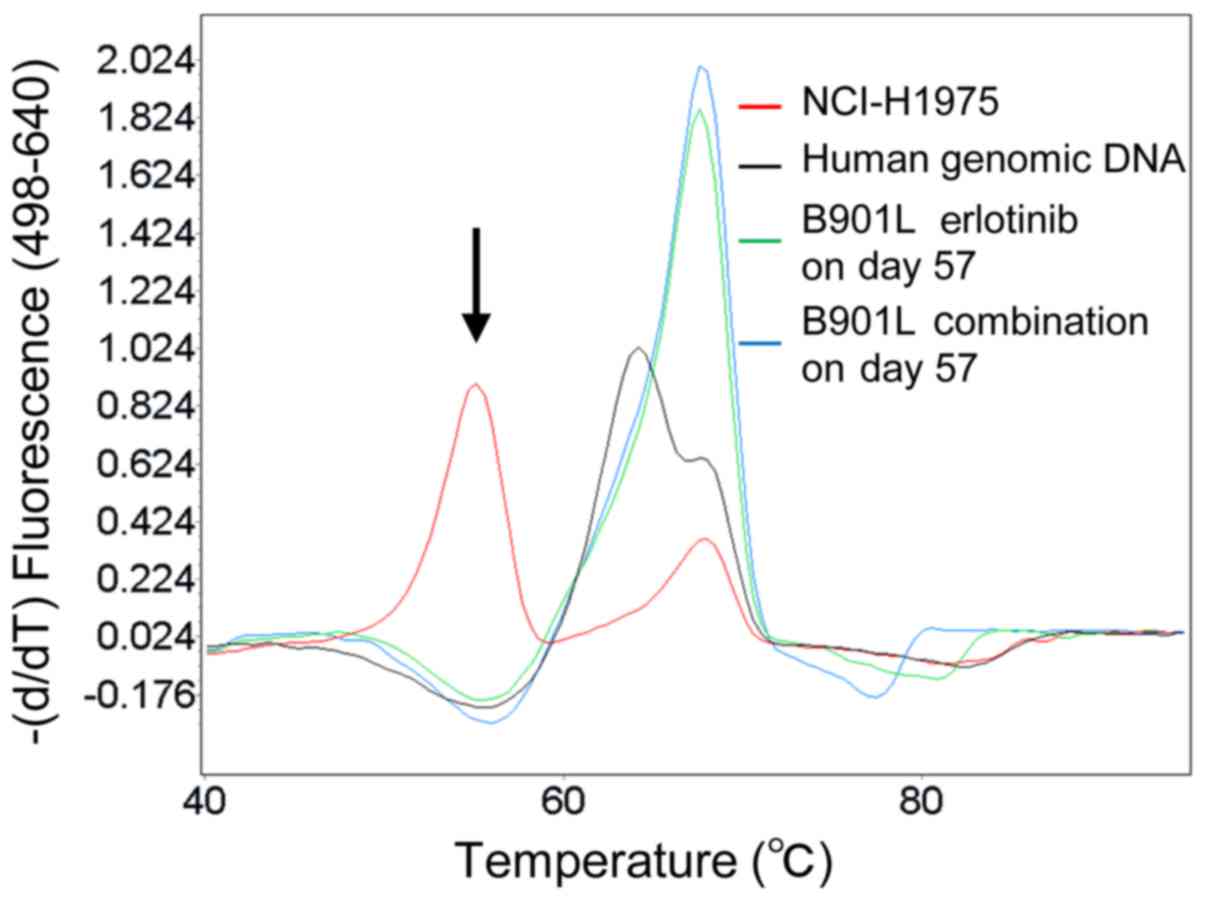
(Fig. 4).
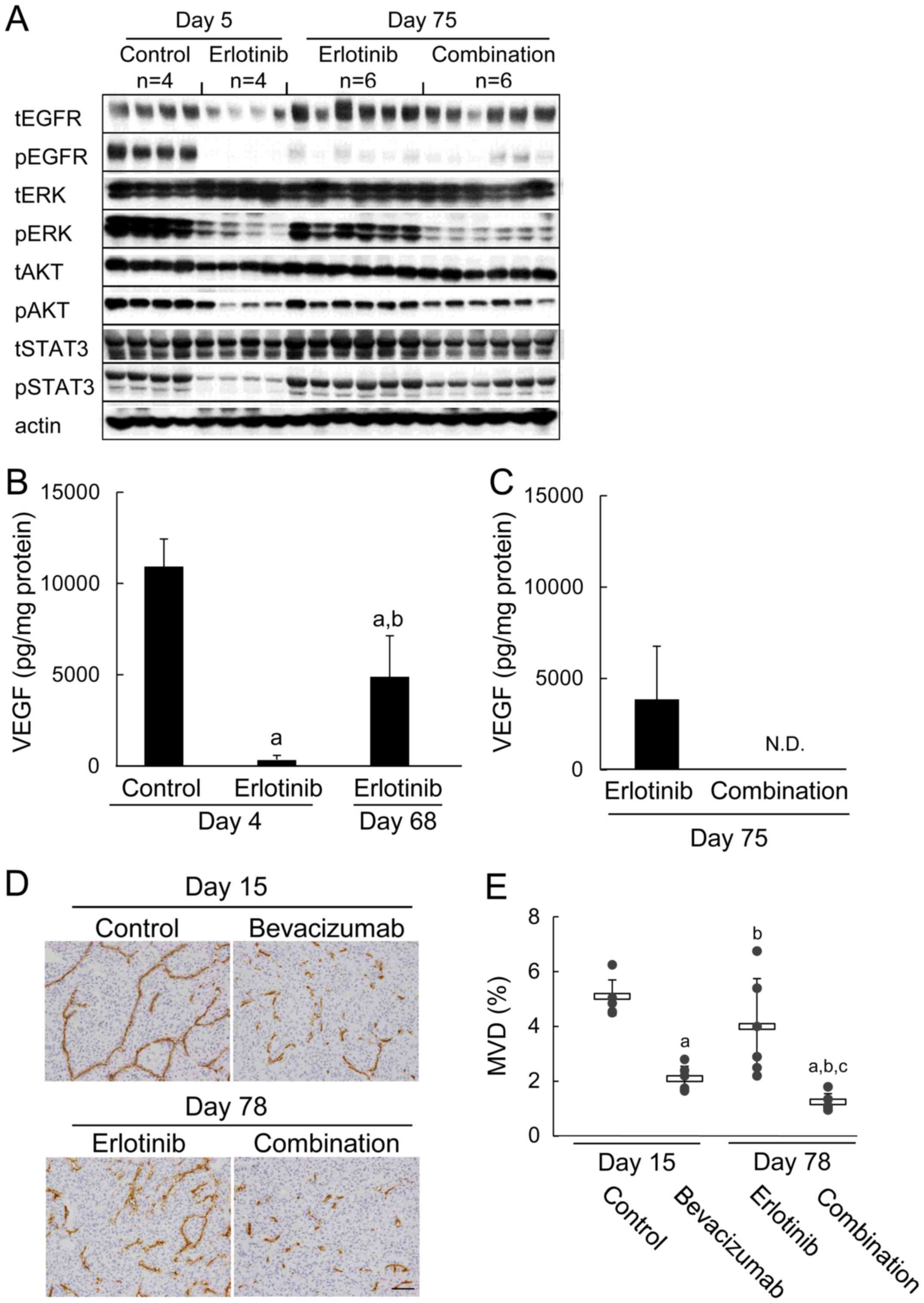 | Figure 3Re-induction of tumor VEGF protein in
the erlotinib-refractory phase, and erlotinib plus
bevacizumab-induced suppression of MVD and signaling pathways. (A)
immunoblots of tumor lysates 3 h after treatment with control, 5
mg/kg of bevacizumab, 60 mg/kg of erlotinib, or erlotinib plus
bevacizumab combination. Tumor samples (n=4 or 6) were collected on
day 5 (control or erlotinib) or day 75 (erlotinib or combination).
Total EGFR (tEGFR), phosphorylated EGFR (pEGFR), total ERK (tERK),
phosphorylated ERK (pERK), total AKT (tAKT), phosphorylated AKT
(pAKT), total STAT3 (tSTAT3), phosphorylated STAT3 (pSTAT3), and
actin were detected by a manual western blotting method. (B) Levels
of VEGF protein expression in tumors after 3 h of treatment with
control or 60 mg/kg of erlotinib. Tumor samples were collected on
day 4 (control or erlotinib) or day 68 (erlotinib) (n=5 or 6). Bars
represent the mean + SD. a, P<0.05 versus control; b, P<0.05
versus day 4 erlotinib (by Wilcoxon test). (C) Tumor samples were
collected on day 75 (erlotinib or combination) (n=4). (D)
Representative immunohistochemical images of CD31-immunostaining to
determine MVD in tumor tissues from mice treated with control, 5
mg/kg of bevacizumab, 60 mg/kg of erlotinib, or combination. Tumor
samples (n=6) were collected on day 15 (control or bevacizumab) or
day 78 (erlotinib or combination). Scale bar is 100 μm. (E)
MVD in the control, bevacizumab, erlotinib, and erlotinib plus
bevacizumab groups. Bars represent mean ± SD. a, P<0.05 versus
control; b, P<0.05 versus bevacizumab; c, P<0.05 versus
erlotinib (by Wilcoxon test). |
Next, we quantified human VEGF concentrations by
using tumor specimens obtained in the erlotinib-sensitive and
erlotinib-refractory phases. In the erlotinib-sensitive phase (day
4), human VEGF concentrations were reduced significantly compared
to concentrations in the control group. Interestingly, human VEGF
levels in the erlotinib-refractory phase (day 68) were increased
(P<0.05) compared with levels in the erlotinib-sensitive phase
(day 4) (Fig. 3B). In contrast,
levels of free human VEGF in the erlotinib plus bevacizumab group
on day 75 were below the detection limit (Fig. 3C). pERK was decreased strongly and
pAKT and pSTAT3 tended to be suppressed in the erlotinib plus
bevacizumab group compared to in the erlotinib group (day 75)
(Fig. 3A).
Then we evaluated MVD in tumor tissues. Significant
suppression of MVD was not observed by erlotinib in the
erlotinib-refractory phase on day 78 compared with control on day
15 (Fig. 3D and E) although MVD
was suppressed by erlotinib on day 4 compared with control in its
sensitive phase (p<0.05) (Fig.
2E). Mean MVD (%) in the bevacizumab group (2.06±0.47) was
inhibited compared with mean MVD in the control group on day 15
(5.04±0.65). Furthermore, MVD was significantly more suppressed by
erlotinib plus bevacizumab on day 78 (1.20±0.32) compared with
bevacizumab on day 15 (Fig. 3D and
E). In contrast to VEGF levels, the levels of bFGF, TGF-α,
G-CSF, and CXCL2 in the erlotinib-refractory phase were not
increased compared with their levels in the erlotinib-sensitive
phase (Table I). Although a
significant increase in IL-6 was observed in the
erlotinib-refractory phase compared to levels in the
erlotinib-sensitive phase, the levels of IL-6 were much lower than
in the control group (Table I).
Taken together, these results suggest that VEGF is involved, at
least in part, in erlotinib resistance mechanisms. Furthermore, the
antiangiogenic effect and the inhibition of signal transduction
were suggested to be mechanisms underlying the antitumor activity
of the combination of erlotinib plus bevacizumab.
Inhibition of VEGF after establishment of
refractoriness to erlotinib showed significant but limited
antitumor efficacy
Since the data described above suggested the
possibility that tumor VEGF, the production of which reappeared
even under the presence of erlotinib, was the key molecule behind
tumor regrowth, we examined the effect of VEGF inhibition by
bevacizumab after the tumors became refractory to erlotinib. After
tumor regrowth was observed, mice were re-randomized on day 64 and
allocated to receive erlotinib, bevacizumab, or the combination of
erlotinib plus bevacizumab. Tumor growth was inhibited by erlotinib
plus bevacizumab compared with erlotinib, although complete tumor
regression was not observed (Fig.
5A). Furthermore, pERK was suppressed and pAKT and pSTAT3
tended to be suppressed by erlotinib plus bevacizumab compared with
erlotinib (Fig. 5B). MVD was also
inhibited by erlotinib plus bevacizumab compared with erlotinib
(Fig. 5C and D).
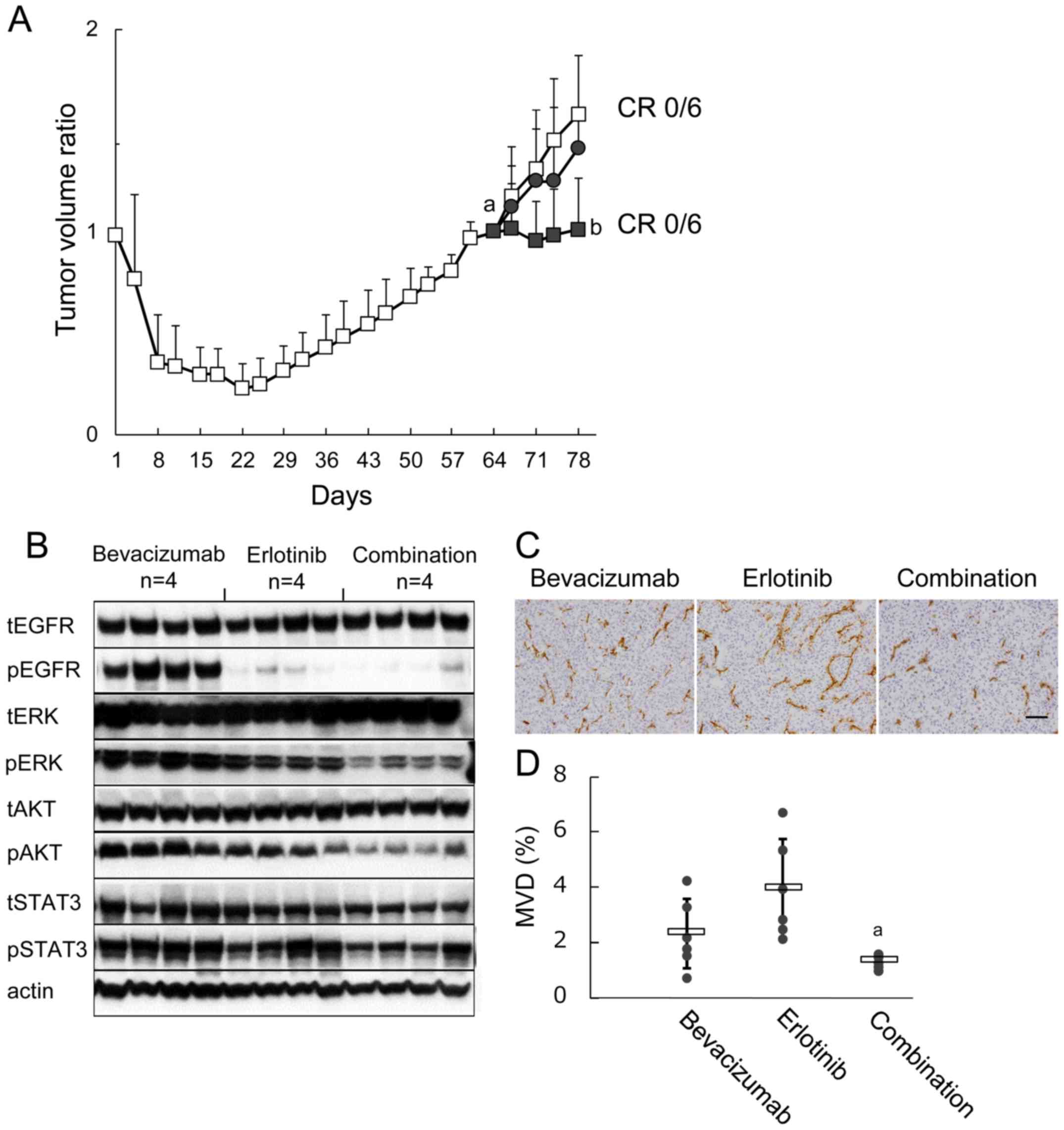 | Figure 5Effect of bevacizumab treatment after
establishing erlotinib refractoriness on signaling pathways, MVD,
and subsequent tumor growth. (A) B901L xenograft-bearing mice were
continuously treated with 60 mg/kg of erlotinib (open squares).
Tumor re-growth was observed during further erlotinib treatment,
and mice were re-randomized into 3 groups on day 64 (n=6). Mice
were treated with 5 mg/kg of bevacizumab (closed circles), 60 mg/kg
of erlotinib (open squares), or combination (closed squares) (n=6).
Each bar represents the mean + SD. a, P<0.05 versus TV ratio of
erlotinib-treated group on day 22; b, P<0.05 versus TV ratio of
erlotinib-treated group on day 78 (Wilcoxon test). (B) Immunoblots
of tumor lysates 3 h after treatment with bevacizumab, erlotinib,
or combination. Tumor samples (n=4) were collected on day 78. Total
EGFR (tEGFR), phosphorylated EGFR (pEGFR), total ERK (tERK),
phosphorylated ERK (pERK), total AKT (tAKT), phosphorylated AKT
(pAKT), total STAT3 (tSTAT3), phosphorylated STAT3 (pSTAT3), and
actin were detected by a manual western blotting method. (C)
Representative immunohistochemical images of CD31-immunostaining to
determine MVD in tumor tissues collected on day 78 (n=6). Scale bar
is 100 μm. (D) MVD in the bevacizumab, erlotinib, and
erlotinib plus bevacizumab groups. Bars represent mean ± SD. (A)
P<0.05 versus erlotinib (by Wilcoxon test). |
Discussion
In the B901L xenograft model, tumor regrowth was
observed following initial strong tumor regression by erlotinib
monotherapy (Fig. 1). In the
erlotinib-sensitive phase, pEGFR and its downstream pERK, pAKT, and
pSTAT3 were suppressed by erlotinib (Fig. 2A), and the expression of tumor VEGF
was decreased significantly compared with control (Fig. 3B) in agreement with previously
reported downregulation of VEGF by EGFR-TKIs (16,17,21).
On the other hand, pERK, pAKT, and pSTAT3 were increased in the
erlotinib-refractory phase compared with that in the
erlotinib-sensitive phase, although pEGFR was still suppressed
(Fig. 3A). The T790M mutation,
which is one of the major erlotinib resistance mechanisms occurring
in the EGFR tyrosine kinase domain and causing a relative decrease
in binding with EGFR-TKIs, was not detected after progression in
this model (Fig. 4). Accordingly,
it is suggested that bypass pathways other than EGFR activate
signaling pathways leading to tumor regrowth. To date, MET
amplification, ERBB2 amplification, and overexpression of
hepatocyte growth factor (HGF), a ligand for MET, K-RAS mutation
and PTEN loss have been proposed as erlotinib resistance mechanisms
(5,22). However, no change in MET or ERBB2
gene expression nor in HGF protein level were observed and K-RAS
mutation was not detected in the tumors in this model (data not
shown). Regarding PTEN loss, the amount of pAKT was significantly
reduced by erlotinib in earlier sensitive phase and it was
increased in refractory phase suggesting activation of PI3K-AKT
pathway. Although PTEN loss was considered to be one of its
mechanisms, pAKT was decreased by addition of bevacizumab even
after acquisition of erlotinib resistance so that we considered it
unlikely that observed increase in pAKT was caused by gene mutation
or deletion such as PTEN loss. On the other hand, tumor VEGF was
markedly restored in the erlotinib-refractory phase, although most
of the VEGF production was suppressed by EGFR during the
erlotinib-sensitive phase (Fig.
3B).
VEGF expression is driven by many factors that are
characteristic of tumors, including oncogene expression, e.g.
ras, src, ERBB2, and EGFR, and hypoxia (18). Under hypoxia, VEGF is principally
regulated by hypoxia-inducible transcription factors (HIFs)
(23). Although the mechanism
leading to VEGF re-induction in the erlotinib-refractory phase has
not yet been fully investigated in this model, in the
erlotinib-sensitive phase, MVD was suppressed significantly by
erlotinib (Fig. 2D and E), at
least in part via downregulation of EGFR-mediated tumor VEGF
production, so there is a possibility that hypoxia in the tumor
induced by erlotinib and subsequent HIF1α activation may lead to
EGFR-independent VEGF expression. On the other hand, besides EGFR,
the HER family has also been indicated as playing a role with
regard to VEGF regulation (23).
For example, monoclonal antibodies targeting HER2 attenuated VEGF
expression (24), while VEGF
production was enhanced in tumor cells exposed to the HER3/HER4
ligand heregulin (25). In the
erlotinib-treated group, pHER2 was activated in the
erlotinib-refractory phase but not in the erlotinib-sensitive phase
in almost all mice (data not shown). Therefore, HER activation and
signaling might also induce VEGF expression.
MVD and signaling pathways were also augmented in
the erlotinib-refractory phase suggesting that VEGF-mediated
angiogenesis and bypass signal activation leads to erlotinib
resistance. Indeed, as shown in Fig.
5, inhibition by bevacizumab of the re-induced VEGF production
after the erlotinib-refractory phase decreased MVD and signal
transduction leading to inhibition of tumor growth. Taken together,
these results indicate that VEGF may be a key molecule in erlotinib
resistance. Association of VEGF production and erlotinib resistance
has also been reported in studies using EGFR wild-type tumor cells.
For example, constitutive VEGF up-regulation in anti-EGFR-resistant
variants of A431 squamous cell carcinoma was reported (26), and erlotinib resistance was
associated with a rise in both tumor cell VEGF and host stromal
VEGF in A549 epithelial carcinoma (27).
Of note, although complete tumor regression was
observed when erlotinib plus bevacizumab was administered from day
1, complete regression was not observed by bevacizumab add-on after
the erlotinib-refractory phase. Furthermore, although there was no
significant difference between TV in the erlotinib group and TV in
the erlotinib plus bevacizumab group in the erlotinib-sensitive
phase, the number of Ki-67+ cells and MVD were decreased
in the erlotinib plus bevacizumab group as compared with in the
erlotinib group (Fig. 2C and E).
Schicher et al showed that bevacizumab decreased the number
of Ki-67+ cells in tumors (28). These results suggest that the
reduction in the number of proliferating cells and MVD in the
tumors, which was caused by co-administration of bevacizumab during
the erlotinib-sensitive phase, led to the inhibition of regrowth.
Furthermore, MVD and pERK were inhibited throughout the study
period in the erlotinib plus bevacizumab group (Figs. 2A and E; 3A and E). Taken together, these results
indicate that co-administration of bevacizumab both in the
erlotinib-sensitive phase and in the erlotinib-refractory phase
achieves maximum efficacy. Although pAKT also tended to be
suppressed by co-administration of bevacizumab in
erlotinib-refractory phase, the level of pAKT was slightly higher
than that in erlotinib-sensitive phase. It was suggested that
unidentified upstream factors besides VEGF contributed to this
modest activation of pAKT.
MVD was suppressed significantly more strongly in
the erlotinib plus bevacizumab group compared with in the group
administered bevacizumab alone (Figs.
2E and 3E). Since erlotinib
has been reported to block production of the angiogenic factors
bFGF and TGF-α in addition to VEGF (18,29),
we investigated if other angiogenic factors besides VEGF were
suppressed with erlotinib. In this model, the levels of bFGF and
TGF-α were not affected by erlotinib and did not increase in the
erlotinib-refractory phase. On the other hand, human G-CSF and
human CXCL2, which were reported to have pro-angiogenic activities
(30–33), were inhibited by erlotinib in
erlotinib-sensitive phase and inhibition of human CXCL2 was
statistically significant until the end of study period (Table I). These results suggested that
EGFR signal is the inducer of pro-angiogenic factors such as CXCL2
production by tumor throughout the study period in contrast to
VEGF, which is induced by other mechanism in erlotinib-refractory
phase.
Inhibition of VEGF by bevacizumab and inhibition of
other angiogenic factors besides VEGF by erlotinib led to the
combination effect on MVD. IL-6 production was greatly reduced by
erlotinib in the erlotinib-sensitive phase, whereas a slight but
statistically significant increase in IL-6 production was observed
in the erlotinib-refractory phase. Since IL-6 is reported to have
pro-angiogenic activity either directly by acting on vascular
endothelial cells or indirectly via STAT3-induced VEGF production
(34,35), the contribution of IL-6 on
angiogenesis as well as tumor cell STAT3 activation in our model
are to be further investigated.
Our findings suggested that VEGF was an inducer of
growth signals through VEGFR- and/or VEGF-mediated production of
tumor/host growth factors in our model. Several studies have
reported that VEGFRs are expressed in many human tumor types and
that VEGF is an autocrine growth factor for tumor cell lines that
express VEGFRs (36–38). On the other hand, tumor cells exist
within a complex microenvironment comprising numerous cells
including, for example, vascular endothelial cells of the blood and
lymphatic circulatory system, stromal fibroblasts, tumor associated
macrophages (TAMs), and myeloid derived suppressor cells (MDSCs).
Furthermore, paracrine signaling interactions between tumor cells
and stromal cells are a key component in the proliferation of
tumors in several organs (39,40).
Cumulative evidence shows that VEGF can induce tumor growth, not
only by promoting angiogenesis but also by creation of other
favorable tumor microenvironments (41). TAMs express VEGFR2, and selective
inhibition of VEGFR2 reduces recruitment of macrophages into
orthotopic pancreatic tumors (42), and treatment with sunitinib, a
receptor tyrosine kinase inhibitor, decreased the number of MDSCs
in advanced tumor-bearing animals (43). A number of
CD11b+Gr-1+ MDSCs and
CD11b+CD206+ macrophages infiltrated into
tumors in our model (data not shown). Therefore, VEGF secreted by
the tumor may activate tumor growth directly or indirectly through
host cells including MDSC and TAM.
In conclusion, continuous treatment with erlotinib
plus bevacizumab shows promising efficacy in the B901L xenograft
model of EGFR Mut+ NSCLC. Furthermore,
re-induction of VEGF and subsequent VEGF-dependent tumor growth is
suggested as one of the major mechanisms of acquired resistance to
erlotinib. Therefore, remarkably prolonged antitumor activity was
achieved by inhibition of VEGF by bevacizumab in combination with
erlotinib. In this study, we established a model that became
refractory to erlotinib after long-term administration of erlotinib
and in which prolonged antitumor activity was shown by treatment
with erlotinib plus bevacizumab, which is in line with the results
of the Phase II trial (JO25567) to evaluate the efficacy of
erlotinib plus bevacizumab in EGFR Mut+ NSCLC
patients. A Phase iii trial (NEJ026) is currently underway. Further
studies are required to better elucidate the mechanisms of action
of erlotinib plus bevacizumab in NSCLC harboring EGFR
mutations.
Glossary
Abbreviations
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
VEGF
|
vascular endothelial cell growth
factor
|
|
VEGFR
|
vascular endothelial cell growth
factor receptor
|
|
TKI
|
tyrosine kinase inhibitor
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
bFGF
|
basic fibroblast growth factor
|
|
TGF-α
|
transforming growth factor-α
|
|
PlGF
|
placental growth factor
|
|
G-CSF
|
granulocyte-colony stimulating
factor
|
|
IL-6
|
interleukin-6
|
|
CXCL2
|
chemokine (C-X-C motif) ligand 2
|
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR Mut+
|
activating EGFR mutations
|
|
PFS
|
progression-free survival
|
|
MVD
|
microvessel density
|
|
HIFs
|
hypoxia-inducible transcription
factors
|
|
TAMs
|
tumor associated macrophages
|
|
MDSCs
|
myeloid derived suppressor cells
|
Acknowledgments
The authors thank Masako Miyazaki, Kumiko Kondoh,
and Hiromi Sawamura at Chugai for their technical assistance with
drug administration, and also thank Dr Yoriko Yamashita-Kashima, Dr
Kazushige Mori, and Dr Kaori Fujimoto-Ouchi for their helpful
support and advice in this study.
References
|
1
|
Shepherd FA, Rodrigues Pereira J, Ciuleanu
T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S,
Smylie M, Martins R, et al National Cancer Institute of Canada
Clinical Trials Group: Erlotinib in previously treated
non-small-cell lung cancer. N Engl J Med. 353:123–132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al Spanish Lung Cancer Group in
collaboration with Groupe Français de Pneumo-Cancérologie and
Associazione Italiana Oncologia Toracica: Erlotinib versus standard
chemotherapy as first-line treatment for European patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomised phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuan F, Chen Y, Dellian M, Safabakhsh N,
Ferrara N and Jain RK: Time-dependent vascular regression and
permeability changes in established human tumor xenografts induced
by an anti-vascular endothelial growth factor/vascular permeability
factor antibody. Proc Natl Acad Sci USA. 93:14765–14770. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Connor JP, Carano RA, Clamp AR, Ross J,
Ho CC, Jackson A, Parker GJ, Rose CJ, Peale FV, Friesenhahn M, et
al: Quantifying antivascular effects of monoclonal antibodies to
vascular endothelial growth factor: Insights from imaging. Clin
Cancer Res. 15:6674–6682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gerber HP and Ferrara N: Pharmacology and
pharmacodynamics of bevacizumab as monotherapy or in combination
with cytotoxic therapy in preclinical studies. Cancer Res.
65:671–680. 2005.PubMed/NCBI
|
|
9
|
Yanagisawa M, Yorozu K, Kurasawa M, Nakano
K, Furugaki K, Yamashita Y, Mori K and Fujimoto-Ouchi K:
Bevacizumab improves the delivery and efficacy of paclitaxel.
Anticancer Drugs. 21:687–694. 2010.PubMed/NCBI
|
|
10
|
Turley RS, Fontanella AN, Padussis JC,
Toshimitsu H, Tokuhisa Y, Cho EH, Hanna G, Beasley GM, Augustine
CK, Dewhirst MW, et al: Bevacizumab-induced alterations in vascular
permeability and drug delivery: A novel approach to augment
regional chemotherapy for in-transit melanoma. Clin Cancer Res.
18:3328–3339. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sandler A, Gray R, Perry MC, Brahmer J,
Schiller JH, Dowlati A, Lilenbaum R and Johnson DH:
Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell
lung cancer. N Engl J Med. 355:2542–2550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herbst RS, Johnson DH, Mininberg E,
Carbone DP, Henderson T, Kim ES, Blumenschein G Jr, Lee JJ, Liu DD,
Truong MT, et al: Phase I/II trial evaluating the anti-vascular
endothelial growth factor monoclonal antibody bevacizumab in
combination with the HER-1/epidermal growth factor receptor
tyrosine kinase inhibitor erlotinib for patients with recurrent
non-small-cell lung cancer. J Clin Oncol. 23:2544–2555. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Camp ER, Summy J, Bauer TW, Liu W, Gallick
GE and Ellis LM: Molecular mechanisms of resistance to therapies
targeting the epidermal growth factor receptor. Clin Cancer Res.
11:397–405. 2005.PubMed/NCBI
|
|
14
|
Seto T, Kato T, Nishio M, Goto K, Atagi S,
Hosomi Y, Yamamoto N, Hida T, Maemondo M, Nakagawa K, et al:
Erlotinib alone or with bevacizumab as first-line therapy in
patients with advanced non-squamous non-small-cell lung cancer
harbouring EGFR mutations (JO25567): An open-label, randomised,
multicentre, phase 2 study. Lancet Oncol. 15:1236–1244. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H, Takayama K, Wang S, Shiraishi Y,
Gotanda K, Harada T, Furuyama K, Iwama E, Ieiri I, Okamoto I, et
al: Addition of bevacizumab enhances antitumor activity of
erlotinib against non-small cell lung cancer xenografts depending
on VEGF expression. Cancer Chemother Pharmacol. 74:1297–1305. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao
GD and Maity A: EGFR tyrosine kinase inhibitors decrease VEGF
expression by both hypoxia-inducible factor (HIF)-1-independent and
HIF-1-dependent mechanisms. Cancer Res. 66:3197–3204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee JG and Wu R: Erlotinib-cisplatin
combination inhibits growth and angiogenesis through c-MYC and
HIF-1α in EGFR-mutated lung cancer in vitro and in vivo. Neoplasia.
17:190–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tabernero J: The role of VEGF and EGFR
inhibition: Implications for combining anti-VEGF and anti-EGFR
agents. Mol Cancer Res. 5:203–220. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chatterjee S, Wieczorek C, Schöttle J,
Siobal M, Hinze Y, Franz T, Florin A, Adamczak J, Heukamp LC,
Neumaier B, et al: Transient antiangiogenic treatment improves
delivery of cytotoxic compounds and therapeutic outcome in lung
cancer. Cancer Res. 74:2816–2824. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Furugaki K, Yasuno H, Iwai T, Moriya Y,
Harada N and Fujimoto-Ouchi K: Melting curve analysis for mutations
of EGFR and KRAS. Anticancer Res. 34:613–621. 2014.PubMed/NCBI
|
|
21
|
Furugaki K, Fukumura J, Iwai T, Yorozu K,
Kurasawa M, Yanagisawa M, Moriya Y, Yamamoto K, Suda K, Mizuuchi H,
et al: Impact of bevacizumab in combination with erlotinib on
EGFR-mutated non-small cell lung cancer xenograft models with T790M
mutation or MET amplification. Int J Cancer. 138:1024–1032. 2016.
View Article : Google Scholar
|
|
22
|
Nakade J, Takeuchi S, Nakagawa T, Ishikawa
D, Sano T, Nanjo S, Yamada T, Ebi H, Zhao L, Yasumoto K, et al:
Triple inhibition of EGFR, Met, and VEGF suppresses regrowth of
HGF-triggered, erlotinib-resistant lung cancer harboring an EGFR
mutation. J Thorac Oncol. 9:775–783. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Larsen AK, Ouaret D, El Ouadrani K and
Petitprez A: Targeting EGFR and VEGF(R) pathway cross-talk in tumor
survival and angiogenesis. Pharmacol Ther. 131:80–90. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Petit AM, Rak J, Hung MC, Rockwell P,
Goldstein N, Fendly B and Kerbel RS: Neutralizing antibodies
against epidermal growth factor and ErbB-2/neu receptor tyrosine
kinases down-regulate vascular endothelial growth factor production
by tumor cells in vitro and in vivo: Angiogenic implications for
signal transduction therapy of solid tumors. Am J Pathol.
151:1523–1530. 1997.PubMed/NCBI
|
|
25
|
Yen L, You XL, Al Moustafa AE, Batist G,
Hynes NE, Mader S, Meloche S and Alaoui-Jamali MA: Heregulin
selectively up regulates vascular endothelial growth factor
secretion in cancer cells and stimulates angiogenesis. Oncogene.
19:3460–3469. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Viloria-Petit A, Crombet T, Jothy S,
Hicklin D, Bohlen P, Schlaeppi JM, Rak J and Kerbel RS: Acquired
resistance to the antitumor effect of epidermal growth factor
receptor-blocking antibodies in vivo: A role for altered tumor
angiogenesis. Cancer Res. 61:5090–5101. 2001.PubMed/NCBI
|
|
27
|
Naumov GN, Nilsson MB, Cascone T, Briggs
A, Straume O, Akslen LA, Lifshits E, Byers LA, Xu L, Wu HK, et al:
Combined vascular endothelial growth factor receptor and epidermal
growth factor receptor (EGFR) blockade inhibits tumor growth in
xenograft models of EGFR inhibitor resistance. Clin Cancer Res.
15:3484–3494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schicher N, Paulitschke V, Swoboda A,
Kunstfeld R, Loewe R, Pilarski P, Pehamberger H and Hoeller C:
Erlotinib and bevacizumab have synergistic activity against
melanoma. Clin Cancer Res. 15:3495–3502. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herbst RS and Sandler A: Bevacizumab and
erlotinib: A promising new approach to the treatment of advanced
NSCLC. Oncologist. 13:1166–1176. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu AX, Duda DG, Sahani DV and Jain RK:
HCC and angiogenesis: Possible targets and future directions. Nat
Rev Clin Oncol. 8:292–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shojaei F, Wu X, Qu X, Kowanetz M, Yu L,
Tan M, Meng YG and Ferrara N: G-CSF-initiated myeloid cell
mobilization and angiogenesis mediate tumor refractoriness to
anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA.
106:6742–6747. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vandercappellen J, Van Damme J and Struyf
S: The role of CXC chemokines and their receptors in cancer. Cancer
Lett. 267:226–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Strieter RM, Burdick MD, Gomperts BN,
Belperio JA and Keane MP: CXC chemokines in angiogenesis. Cytokine
Growth Factor Rev. 16:593–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB,
Lee CN and Hsieh CY: Interleukin-6 promotes cervical tumor growth
by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene.
22:1517–1527. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nilsson MB, Langley RR and Fidler IJ:
Interleukin-6, secreted by human ovarian carcinoma cells, is a
potent proangiogenic cytokine. Cancer Res. 65:10794–10800. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barr MP, Gray SG, Gately K, Hams E, Fallon
PG, Davies AM, Richard DJ, Pidgeon GP and O'Byrne KJ: Vascular
endothelial growth factor is an autocrine growth factor, signaling
through neuropilin-1 in non-small cell lung cancer. Mol Cancer.
14:452015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Masood R, Cai J, Zheng T, Smith DL, Hinton
DR and Gill PS: Vascular endothelial growth factor (VEGF) is an
autocrine growth factor for VEGF receptor-positive human tumors.
Blood. 98:1904–1913. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Murdoch C, Muthana M, Coffelt SB and Lewis
CE: The role of myeloid cells in the promotion of tumour
angiogenesis. Nat Rev Cancer. 8:618–631. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Voron T, Marcheteau E, Pernot S, Colussi
O, Tartour E, Taieb J and Terme M: Control of the immune response
by pro-angiogenic factors. Front Oncol. 4:702014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dineen SP, Lynn KD, Holloway SE, Miller
AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB and
Brekken RA: Vascular endothelial growth factor receptor 2 mediates
macrophage infiltration into orthotopic pancreatic tumors in mice.
Cancer Res. 68:4340–4346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck
M, Sung M, Schwartz M, Divino CM, Pan PY and Chen SH: The novel
role of tyrosine kinase inhibitor in the reversal of immune
suppression and modulation of tumor microenvironment for
immune-based cancer therapies. Cancer Res. 69:2514–2522. 2009.
View Article : Google Scholar : PubMed/NCBI
|