1. Introduction
As first identified by Polyak et al in 1997
(1), the gene of
lipopolysaccharide (LPS)-induced tumor necrosis factor (TNF)-α
factor (LITAF) was initially termed p53-inducible gene
7 (PIG7), due to the fact that it encodes for a protein
that is positively regulated by the tumor suppressor protein, p53
(1). Two years later, Myokai et
al (2) cloned an LPS-regulated
gene with the same sequence as PIG7. This gene was
subsequently termed LITAF as its encoded protein product,
LITAF, translocated into the nucleus following cellular activation
by LPS, which was followed by the upregulation of TNF-α
transcription (2–4).
It is widely accepted that tumor-associated
inflammation is a major contributor to cancer progression, and it
has been recognized as the seventh hallmark of cancer (5,6).
Numerous primary inflammatory mediators have been identified,
including interleukin (IL)-4 (7),
CCL18 (8) and granulocyte
macrophage colony-stimulating factor (9). Previous observations suggest that
LITAF, as a ubiquitously expressed gene (1–4), may
be an enhancer of inflammatory diseases, as well as a suppressor of
cancer-associated inflammation. In the current review, the
above-mentioned observations are summarized, and LITAF is
presented as a potential novel target for cancer therapy.
2. Structure and general features of
LITAF
Human LITAF is located on chromosome 16 and
it encodes a full length cDNA of 1,551 base pairs (bp), which
contain three major structural components: A 5′ untranslated region
(UTR) of 1,001 bp, 3′ UTR of 76 bp and an open reading frame of 474
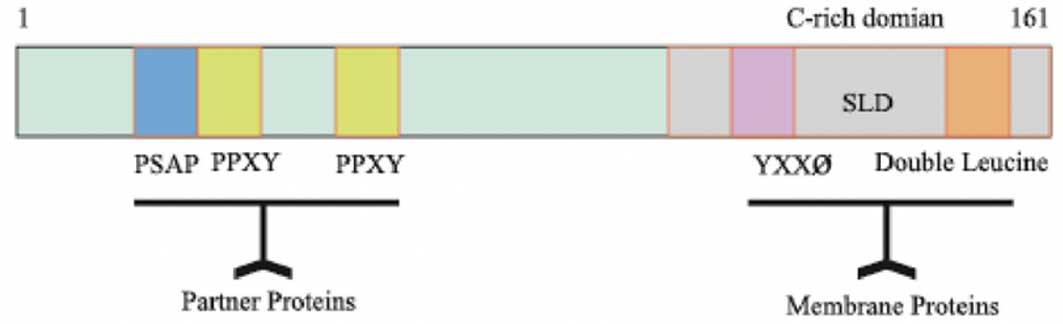
bp (2,10). The C-terminal of the LITAF protein
has enriched cysteine residues and includes a highly conserved C3H4
zinc finger region that is interrupted by 23 hydrophobic amino
acids, called small integral membrane protein of lysosome/late
endosome (SIMPLE)-like domain (SLD) (11). The SLD domain contains a YXX ø (ø
is a hydrophobic amino acid) and double leucine motifs (12). It was reported that proteins
containing the YXX ø motif interact with the clathrin adaptor
compound and are, therefore, able to mediate the import and export
of membrane proteins in the endosome, Golgi apparatus and lysosomes
(13,14). Furthermore, proteins with double
leucine motifs are able to target lysosomes and endosomes (15). However, the N-terminal of the LITAF
protein is enriched with proline residues and has PPXY and PS/TAP
motifs, which mediate the association of LITAF with partner
proteins (16–18) (Fig.
1).
3. Trafficking of LITAF
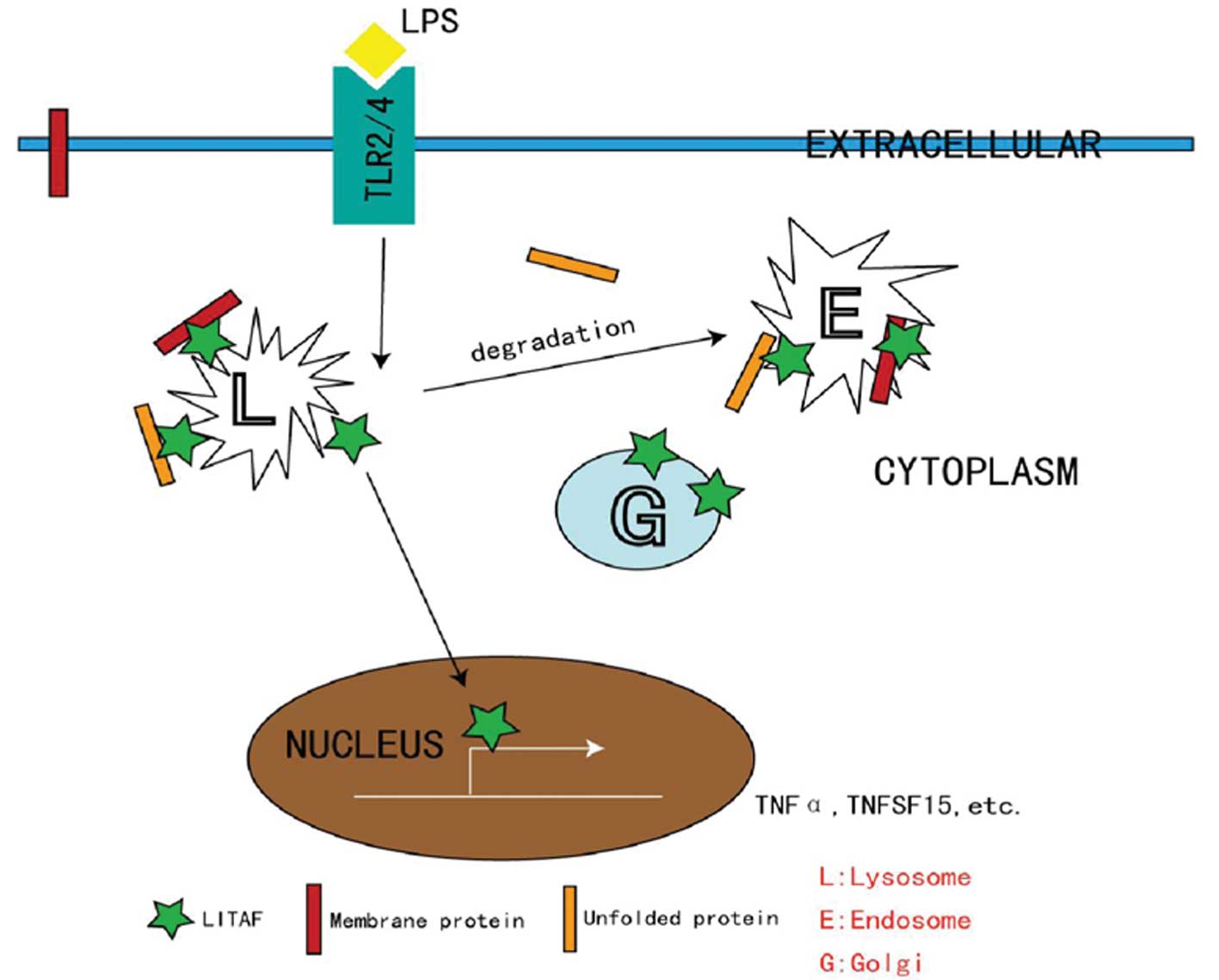
The nuclear translocation and transcription factor
activity of LITAF are critical for the activation of numerous
immune cells via classical pathways (Fig. 2). While intracellular LITAF is
located in the membranes of late endosomes and lysosomes under
quiescent conditions, these processes require free LITAF to be
released from these intracellular compartments. It has been
proposed that such a process is orchestrated by the protein-protein
interactions with ubiquitination-associated proteins, such as the
E3 ligase NEDD4 (16). LITAF
functions with the endosomal sorting complex required for transport
components to control endosome-to-lysosome trafficking (17). As a negative control, previous
studies have indicated that mutated LITAF proteins mislocalize to
the cytosol (18) and/or
mitochondria (19), where they
cease their wild-type (WT) activities and serve as an etiological
cause of Charcot-Marie-Tooth disease, a severe peripheral nervous
system disorder (20,21).
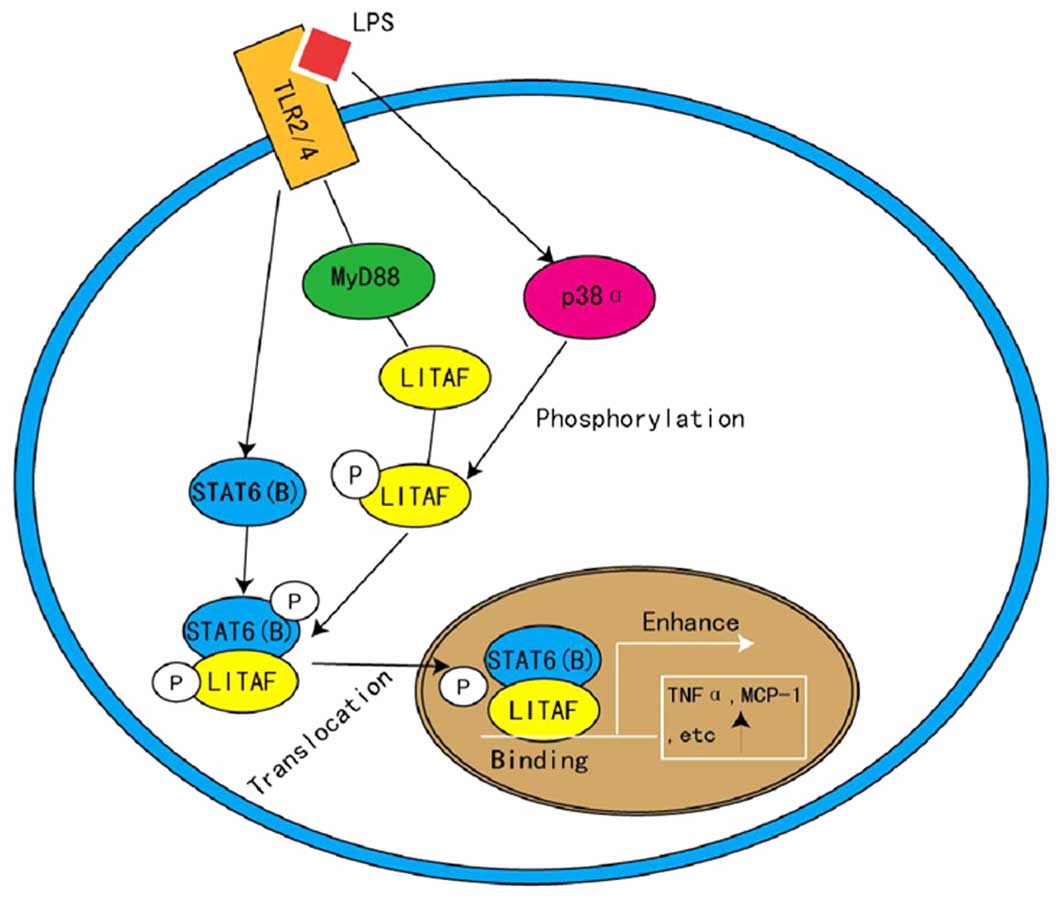
4. LITAF and STAT6 (B) in inflammation
LITAF is known as a TNF-α inducer (22), therefore, it is notable that
transient transfection of LITAF resulted in no significant
elevation of TNF-α levels following LPS treatment (3). This indicates that LPS activates
additional factors, other than LITAF, that also regulate the
transcription of TNF-α and that these factors may be binding
partners of LITAF. Using a yeast two-hybrid system, a transcription
factor, signal transducer and activator of transcription (STAT)6
(B), has been identified as a functional binding partner of LITAF
(3). LITAF and STAT6 (B) are
activated by LPS, then associate with toll-like receptor-2/4 to
form a complex, which is dependent on MyD88 and is phosphorylated
by p38-α (3). Phosphorylated LITAF
and STAT6 (B) consequently interact to form a protein complex prior
to translocating into the nucleus, where LITAF binds specifically
to the promoter sequence, thus activating the expression of
downstream genes, such as TNF-α and IL-6 (4,23)
(Fig. 3). Focusing on this
pathway, LITAF has become a novel target for the treatment of
endotoxic shock and inflammation (24), as implicated by Matsuno et
al (25) who demonstrated that
LITAF-knockout mice were more resistant to LPS-induced
mortality.
5. LITAF and inflammatory diseases
As a significant disease associated with LITAF,
inflammatory bowel disease (IBD) is a type of chronic intestinal
inflammatory disease with an unknown etiology, which includes
ulcerative colitis (UC) and Crohn's disease (CD) (26). The typical pathogenesis of IBD
includes aberrant expression of bowel-specific proinflammatory
cytokines, including TNF-α (24,27,28).
This indicates that LITAF may be involved in IBD and may be
abnormally expressed in this disease. Stucchi et al
(29) observed that the mRNA
levels of LITAF in colon tissue samples from patients with
CD were five times higher than those from healthy controls. In
addition, within the same CD sample, the inflammatory areas
presented with 60% more LITAF mRNA than the non-inflammatory
areas (29). Similar phenomena
have been observed in patients with UC. Colon tissues from patients
with UC expressed LITAF mRNA levels 15 times greater than
healthy individuals (26).
However, in such patients, there was no significant difference in
the mRNA level of LITAF between the inflammatory areas and
the surrounding normal tissues. Immunohistochemistry has
demonstrated that LITAF is predominantly expressed by lamina
propria macrophages (LPM) (29).
This was verified by Bushell et al (30) with a 2,4,6-trinitrobenzene sulfonic
acid (TNBS)-induced mouse colon inflammation model. This study
additionally indicated that mRNA and protein levels of LITAF
were dramatically upregulated in TNBS-treated mice when compared
with untreated mice. Furthermore, the expression of TNF-α in
the LPM from LITAF mac−/− mice was significantly
lower than that of the WT mice (30). These results strongly suggest that
LITAF upregulates expression of TNF-α in LPM and elevated
expression of LITAF coincides with the progression of
IBD.
Arthritis is an inflammatory disease occurring in
the joints of the human body and surrounding tissues, which has a
complex etiology. Causal factors include chronic inflammation,
autoimmune reactions, infection, metabolic disorders, trauma and
degenerative disorders (31).
Patients with arthritis commonly exhibit vascular endothelial
dysfunction with alterations in numerous inflammatory factors,
including TNF-α, IL-6 and IL-8 (32,33).
To investigate whether LITAF was involved in arthritis, Merrill
et al (34) established an
LITAF knockout mouse [tamLITAF(i)−/−] through
tamoxifen induction. LPS was used to treat WT and
tamLITAF(i)−/− mice and collagen-induced arthritis
experiments were performed. The degree of disease severity was
found to be dramatically higher in the WT mice than in the
tamLITAF(i)−/− mice, this observation was noted from 3
days post-treatment and the difference became more significant over
time. In addition, pannus and synovitis inflammations were observed
to be elevated in the tamLITAF(i)−/− mice. Additionally,
the degree of bone resorption was observed to be lower in
tamLITAF(i)−/− mice compared with the WT mice (34). These results suggest that in
vivo depletion of LITAF effectively reduces the harmful
effects of arthritis. Corroborating these results, Srinivasan et
al (35) identified a
connection between LITAF and arthritis, and proposed that it may
involve extracellular-related kinase 1/2 and protein kinase B
(35). These observations suggest
that LITAF may promote the progression of arthritis, as well as
additional associated whole body inflammation in mice.
6. LITAF and cancer
In addition to inflammation, LITAF has been
identified as a potential tumor suppressor gene, due to the fact
that its expression can be induced by p53 (1). Evidence from cohort studies has
revealed that LITAF expression is significantly lower in tumor
tissues when compared with isogenic normal tissues (36,37).
However, the functional mechanisms of the action of LITAF in tumors
remains unclear.
Zhou et al (38) used small hairpin (sh)RNA to disrupt
gene expression in the adenosine monophosphate-activated protein
kinase (AMPK)-LITAF-TNF superfamily member 15 (TNFSF15) signaling
pathway in prostatic cancer cells and elucidated that shRNA
targeting of LITAF (shRNA-LITAF) significantly
enhanced the degree of malignancy of cancer cells. Notably, its
effect was more marked than that of shRNA-p53 (38). Furthermore, Zhou et al
(38) established an allograft
prostatic tumor model by subcutaneous injection of prostatic cancer
cells into nude mice. Following development of tumors, those
analyzed from the shRNA-LITAF group were observed to be
significantly larger in size and weight compared with the tumors
from the shRNA-control group (38). These results suggest that LITAF
inhibits the proliferation of prostatic cancer cells, which
supports the assumption that LITAF functions as a tumor
suppressor gene.
Furthermore, a breast cancer study analyzed the gene
expression of normal breast tissues, ductal carcinoma in
situ (DCIS) and invasive ductal carcinoma (IDC) using the
Serial analysis of gene expression method. The study revealed that
LITAF expression was 29 times lower in DCIS compared with
that of normal tissues, while there was no clear alteration in the
LITAF levels observed in IDC (36). Similarly, Fernandez-Cobo et
al (39) confirmed that
LITAF expression in breast cancer cells was 37 times lower
than that in normal breast epithelial cells. It was hypothesized
that LITAF and other cytokines participate in the recovery process
of breast tissues following pregnancy and lactation, during which
extensive apoptotic events occur in breast tissues (40). Furthermore, lower expression of
LITAF may promote the early transformation of breast tissues by
slowing down the normal apoptotic process.
Wang et al (37) conducted qualitative polymerase
chain reaction analysis and established that bone marrow
LITAF expression in patients with acute leukemia (as well as
refractory and relapsed acute leukemia) is significantly reduced,
when compared with the expression levels in patients at initial
diagnosis. In addition, Wang et al elucidated that the
transient expression of LITAF has little apparent influence
on the proliferation of acute leukemia cells. However, LITAF
markedly enhances the inhibitory effects of etoposide and
daunomycin on acute leukemia, suggesting that LITAF sensitizes
leukemic cells to chemotherapeutic agents (37).
It should be noted that not all cancer cells exhibit
low expression of LITAF. For example, Matsumura et al
(41) examined a rare malignant
skin tumor, extra-mammary Paget's disease (EMPD) and observed that
EMPD tissues exhibited higher expression levels of LITAF in
comparison with isogenic normal tissues, in three out of four
individuals (41). This phenomenon
may be relevant to somatic mutations. The study also identified
LITAF site mutations in three out of 12 cases, among which
two exhibited non-synonymous mutations and one exhibited synonymous
mutations (41). The mechanism of
this mutation and the associated expression remains unclear.
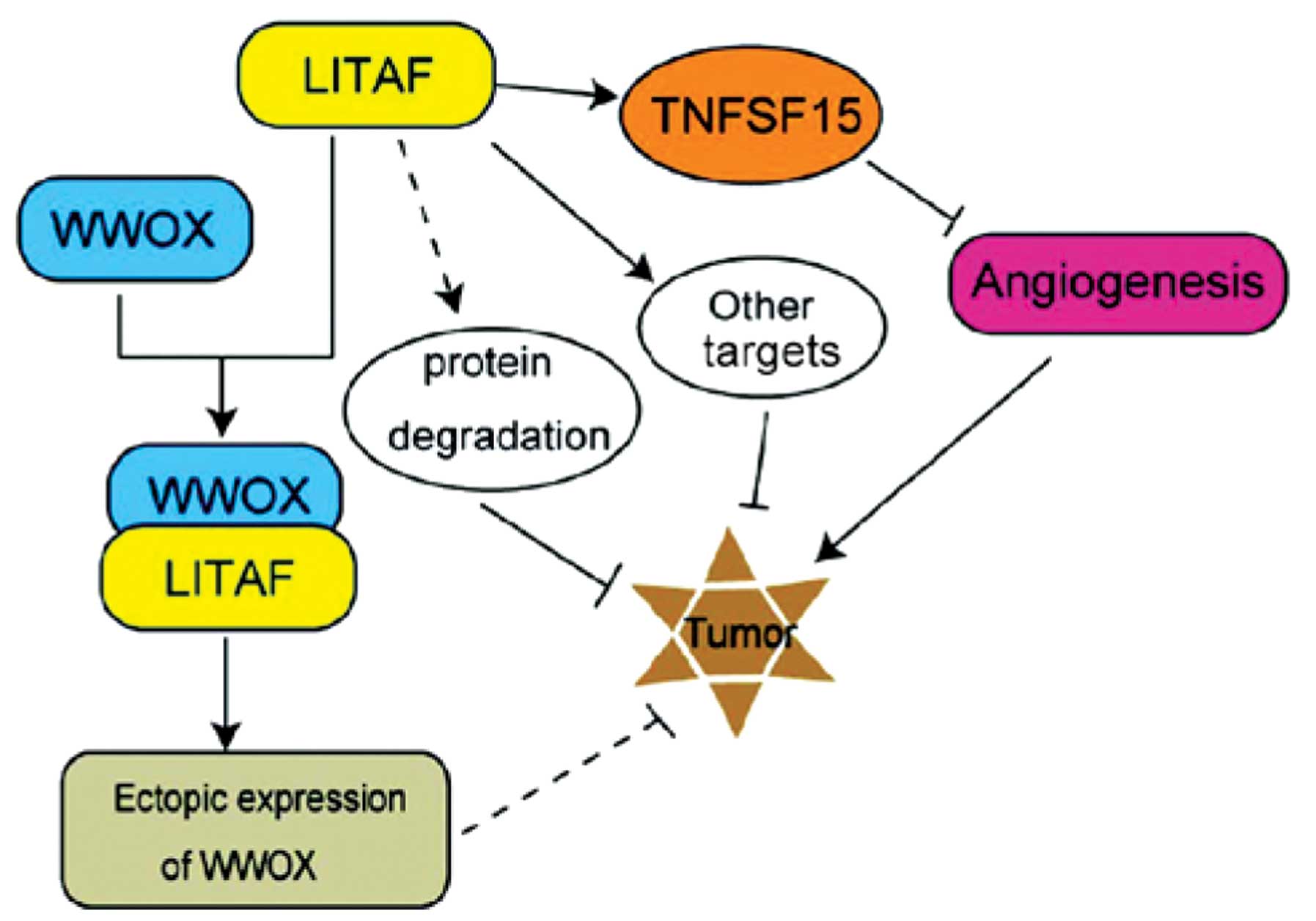
There are numerous mechanisms suggested to be
involved in the tumor suppressor activity of LITAF (Fig. 4). Firstly, the two PPXY motifs at
the N-terminal of LITAF associates with the WW domain containing
proteins, such as NEDD4 and Itch, which are able to promote p53-
and/or p72-mediated cell apoptosis and subsequently restrict tumor
growth (40,42,43).
Secondly, LITAF may promote the ubiquitin-proteasome system in
mediating the degradation of pro-cancerous proteins (44). Thirdly, LITAF is able to stimulate
the expression of TNFSF15 and then restrain angiogenesis to inhibit
tumor growth, as it acts as a downstream target of the tumor
suppressor factor, AMPK (38).
It is hypothesized that LITAF may serve as a switch
in the balance between classical inflammation and alternative
activation in cancer. Immune cell infiltration is a typical trigger
of cancer-associated inflammation. Notably, studies using mouse
models suggested that the alleviation of immune responses results
in a decline in the quantity and size of tumors in the murine body
(45,46). Alternative activation of various
cell types, including tumor-associated macrophages (47), cancer-related fibroblasts (48) and aberrantly activated neutrophils
(49) have been identified in
numerous types of cancer, including breast (50) and colorectal cancer (51), and melanoma (52). In the context of these types of
cancer, the regulators and determinants of classical and
alternative immune activation remain unclear. It has been observed
that LITAF is highly expressed in macrophages in various
acute inflammatory tissues, and classically induces TNF-α, which
exerts antiviral, antitumor and proapoptotic activities when at
sufficiently high in situ concentrations (53,54).
Short-term activation of LITAF inhibits the growth of cancer cells
potentially through proinflammatory effects that target the
expansion of tumor-antigen specific T cells and the cancer cells
themselves (55). During chronic
inflammation, inflammatory factors overexpressed by the alternative
activated immune cells may suppress the expression of LITAF
via the negative feedback mechanism, for example via the nitric
oxide pathway (56). However, the
exact role of LITAF in the transition from inflammation to tumor
suppression requires further investigation, which may elucidate the
potential for LITAF manipulation to modulate early
carcinogenesis and/or cancer progression.
7. Summary and prospect
LITAF may affect cellular functions by either acting
as a transcription factor in mediating target gene expression, or
by acting as a recruiting factor that targets partner proteins to
the lysosome for degradation. Current evidence indicates that
various possible mechanisms may explain the contribution of altered
LITAF expression to the progression of diseases, such as
inflammation or tumors: i) Cytokine levels are dysregulated; ii)
p53-mediated cell apoptosis signaling is affected; iii) Protein
degradation in the lysosome is interrupted. It is proposed that
LITAF may serve as a switch in the balance of classical and
alternative activation in the tumor microenvironment. It remains
unclear whether LITAF is a cause or effect of tumor
inflammation, thus it is an important focus for further
investigation and may be a promising therapeutic target.
Acknowledgments
The current study was supported in part by grants
from The National Science Foundation of China (grant nos. 81171952,
8127292, 31460304 and 81460374) and a grant from Jiangxi Provincial
Department of Science and Technology (grant no. 20133BBG70061). The
authors would also like to thank Dr Zhijun Luo and Dr Yong Xie for
their support.
References
|
1
|
Polyak K, Xia Y, Zweier JL, Kinzler KW and
Vogelstein B: A model for p53-induced apoptosis. Nature.
389:300–305. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Myokai F, Takashiba S, Lebo R and Amar S:
A novel lipopolysaccharide-induced transcription factor regulating
tumor necrosis factor alpha gene expression: Molecular cloning,
sequencing, characterization and chromosomal assignment. Proc Natl
Acad Sci USA. 96:4518–4523. 1999. View Article : Google Scholar
|
|
3
|
Tang X, Marciano DL, Leeman SE and Amar S:
LPS induces the interaction of a transcription factor, LPS-induced
TNF-alpha factor and STAT6 (B) with effects on multiple cytokines.
Proc Natl Acad Sci USA. 102:5132–5137. 2005. View Article : Google Scholar
|
|
4
|
Tang X, Metzger D, Leeman S and Amar S:
LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced
LPS-induced cytokine: Evidence for LITAF-dependent LPS signaling
pathways. Proc Natl Acad Sci USA. 103:13777–13782. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mantovani A: Cancer: Inflaming metastasis.
Nature. 457:36–37. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gocheva V, Wang H-W, Gadea BB, Shree T,
Hunter KE, Garfall AL, Berman T and Joyce JA: IL-4 induces
cathepsin protease activity in tumor-associated macrophages to
promote cancer growth and invasion. Genes Dev. 24:241–255. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen J, Yao Y, Gong C, Yu F, Su S, Chen J,
Liu B, Deng H, Wang F, Lin L, et al: CCL18 from tumor-associated
macrophages promotes breast cancer metastasis via PITPNM3. Cancer
Cell. 19:541–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su S, Liu Q, Chen J, Chen J, Chen F, He C,
Huang D, Wu W, Lin L, Huang W, et al: A Positive Feedback Loop
between mesenchymal-like cancer cells and macrophages is essential
to breast cancer metastasis. Cancer cell. 25:605–620. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bolcato-Bellemin AL, Mattei MG, Fenton M
and Amar S: Molecular cloning and characterization of mouse LITAF
cDNA: role in the regulation of tumor necrosis factor-alpha
(TNF-alpha) gene expression. J Endotoxin Res. 10:15–23.
2004.PubMed/NCBI
|
|
11
|
Moriwaki Y, Begum NA, Kobayashi M,
Matsumoto M, Toyoshima K and Seya T: Mycobacterium bovis Bacillus
Calmette-Guerin and its cell wall complex induce a novel lysosomal
membrane protein, SIMPLE, that bridges the missing link between
lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and
estrogen-inducible gene, EET-1. J Biol Chem. 276:23065–23076. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boge M, Wyss S, Bonifacino JS and Thali M:
A membrane-proximal tyrosine-based signal mediates internalization
of the HIV-1 envelope glycoprotein via interaction with the AP-2
clathrin adaptor. J Biol Chem. 273:15773–15778. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bonifacino JS and Dell'Angelica EC:
Molecular bases for the recognition of tyrosine-based sorting
signals. J Cell Biol. 145:923–926. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simmen T, Schmidt A, Hunziker W and
Beermann F: The tyrosinase tail mediates sorting to the lysosomal
compartment in MDCK cells via a di-leucine and a tyrosine-based
signal. J Cell Sci. 112:45–53. 1999.
|
|
15
|
Letourneur F and Klausner RD: A novel
di-leucine motif and a tyrosine-based motif independently mediate
lysosomal targeting and endocytosis of CD3 chains. Cell.
69:1143–1157. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shirk AJ, Anderson SK, Hashemi SH, Chance
PF and Bennett CL: SIMPLE interacts with NEDD4 and TSG101: Evidence
for a role in lysosomal sorting and implications for
Charcot-Marie-Tooth disease. J Neurosci Res. 82:43–50. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SM, Chin LS and Li L:
Charcot-Marie-Tooth disease-linked protein SIMPLE functions with
the ESCRT machinery in endosomal trafficking. J Cell Biol.
199:799–816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee SM, Olzmann JA, Chin LS and Li L:
Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE
protein mislocalization and degradation by the proteasome and
aggresome-autophagy pathways. J Cell Sci. 124:3319–3331. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferreira Lacerda AF, Hartjes E and
Brunetti CR: LITAF mutations associated with Charcot-Marie-Tooth
Disease 1C Show mislocalization from the late endosome/lysosome to
the mitochondria. PLoS One. 9:e1034542014. View Article : Google Scholar :
|
|
20
|
Ciotti P, Luigetti M, Geroldi A, Capponi
S, Pezzini I, Gulli R, Pazzaglia C, Padua L, Massa R, Mandich P, et
al: A novel LITAF/SIMPLE mutation within a family with a
demyelinating form of Charcot-Marie-Tooth disease. J Neurol Sci.
343:183–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luigetti M, Fabrizi GM, Taioli F, Del
Grande A and Lo Monaco M: A novel LITAF/SIMPLE variant within a
family with minimal demyelinating Charcot-Marie-Tooth disease.
Neurol Sci. 35:2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang X, Molina M and Amar S: p53 short
peptide (p53pep164) regulates lipopolysaccharide-induced tumor
necrosis factor-alpha factor/cytokine expression. Cancer Res.
67:1308–1316. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang X, Woodward T and Amar S: A PTP4A3
peptide PIMAP39 modulates TNF-alpha levels and endotoxic shock. J
Innate Immun. 2:43–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brannigan AE, Watson RW, Beddy D, Hurley
H, Fitzpatrick JM and O'Connell PR: Increased adhesion molecule
expression in serosal fibroblasts isolated from patients with
inflammatory bowel disease is secondary to inflammation. Ann Surg.
235:507–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsuno H, Yudoh K, Katayama R, Nakazawa
F, Uzuki M, Sawai T, Yonezawa T, Saeki Y, Panayi GS, Pitzalis C, et
al: The role of TNF-alpha in the pathogenesis of inflammation and
joint destruction in rheumatoid arthritis (RA): A study using a
human RA/SCID mouse chimera. Rheumatology (Oxford). 41:329–337.
2002. View Article : Google Scholar
|
|
26
|
Stucchi A, Reed K, O'Brien M, Cerda S,
Andrews C, Gower A, Bushell K, Amar S, Leeman S and Becker J: A new
transcription factor that regulates TNF-alpha gene expression,
LITAF, is increased in intestinal tissues from patients with CD and
UC. Inflamm Bowel Dis. 12:581–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baker DA, Barth J, Chang R, Obeid LM and
Gilkeson GS: Genetic sphingosine kinase 1 deficiency significantly
decreases synovial inflammation and joint erosions in murine
TNF-alpha-induced arthritis. J Immunol. 185:2570–2579. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bushell KN, Leeman SE, Gillespie E, Gower
AC, Reed KL, Stucchi AF, Becker JM and Amar S: LITAF mediation of
increased TNF-α secretion from inflamed colonic lamina propria
macrophages. PLoS One. 6:e258492011. View Article : Google Scholar
|
|
29
|
Stucchi A, Reed K, O'Brien M, Cerda S,
Andrews C, Gower A, Bushell K, Amar S, Leeman S and Becker J: A new
transcription factor that regulates TNF-alpha gene expression,
LITAF, is increased in intestinal tissues from patients with CD and
UC. Inflamm Bowel Dis. 12:581–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bushell KN, Leeman SE, Amar S, Reed KL,
Gower AC, Stucchi AF and Becker JM: Macrophage-specific LITAF
(lipopolysaccharide induced TNF-alpha factor) knockout mice (LITAF
mac−/−) have a reduced inflammatory response to colonic
administration of trinitrobenzene sulfonic acid (TNBS). FASEB J.
22(Meeting Abstract Supplement): 1138.42008.
|
|
31
|
Zhang H, Hilton MJ, Anolik JH, Welle SL,
Zhao C, Yao Z, Li X, Wang Z, Boyce BF and Xing L: NOTCH inhibits
osteoblast formation in inflammatory arthritis via noncanonical
NF-κB. J Clin Invest. 124:3200–3214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feldmann M, Brennan FM and Maini RN: Role
of cytokines in rheumatoid arthritis. Annu Rev Immunol. 14:397–440.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brennan FM and McInnes IB: Evidence that
cytokines play a role in rheumatoid arthritis. J Clin Invest.
118:3537–3545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Merrill JC, You J, Constable C, Leeman SE
and Amar S: Whole-body deletion of LPS-induced TNF-α factor (LITAF)
markedly improves experimental endotoxic shock and inflammatory
arthritis. Proc Natl Acad Sci USA. 108:21247–21252. 2011.
View Article : Google Scholar
|
|
35
|
Srinivasan S, Leeman SE and Amar S:
Beneficial dysregulation of the time course of inflammatory
mediators in lipopolysaccharide-induced tumor necrosis factor alpha
factor-deficient mice. Clin Vaccine Immunol. 17:699–704. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abba MC, Drake JA, Hawkins KA, Hu Y, Sun
H, Notcovich C, Gaddis S, Sahin A, Baggerly K and Aldaz CM:
Transcriptomic changes in human breast cancer progression as
determined by serial analysis of gene expression. Breast Cancer
Res. 6:R499–R513. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang D, Liu J, Tang K, Xu Z, Xiong X, Rao
Q, Wang M and Wang J: Expression of pig7 gene in acute leukemia and
its potential to modulate the chemosensitivity of leukemic cells.
Leuk Res. 33:28–38. 2009. View Article : Google Scholar
|
|
38
|
Zhou J, Yang Z, Tsuji T, Gong J, Xie J,
Chen C, Li W, Amar S and Luo Z: LITAF and TNFSF15, two downstream
targets of AMPK, exert inhibitory effects on tumor growth.
Oncogene. 30:1892–1900. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fernandez-Cobo M, Holland JF and Pogo BG:
Transcription profiles of non-immortalized breast cancer cell
lines. BMC Cancer. 6:992006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ludes-Meyers JH, Kil H, Bednarek AK, Drake
J, Bedford MT and Aldaz CM: WWOX binds the specific proline-rich
ligand PPXY: identification of candidate interacting proteins.
Oncogene. 23:5049–5055. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsumura Y, Matsumura Y, Nishigori C,
Horio T and Miyachi Y: PIG7/LITAF gene mutation and overexpression
of its gene product in extramammary Paget's disease. Int J Cancer.
111:218–223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takeuchi T, Adachi Y and Nagayama T: A
WWOX-binding molecule, transmembrane protein 207, is related to the
invasiveness of gastric signet-ring cell carcinoma. Carcinogenesis.
33:548–554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eaton HE, Metcalf J, Lacerda AF and
Brunetti CR: Accumulation of endogenous LITAF in aggresomes. PLoS
One. 7:e300032012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Eaton HE, Desrochers G, Drory SB, Metcalf
J, Angers A and Brunetti CR: SIMPLE/LITAF expression induces the
translocation of the ubiquitin ligase itch towards the lysosomal
compartments. PLoS One. 6:e168732011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van Kempen LC, de Visser KE and Coussens
LM: Inflammation, proteases and cancer. Eur J Cancer. 42:728–734.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
de Visser KE and Coussens LM: The
inflammatory tumor microenvironment and its impact on cancer
development. Contrib Microbiol. 13:118–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mantovani A, Schioppa T, Porta C, Allavena
P and Sica A: Role of tumor-associated macrophages in tumor
progression and invasion. Cancer Metastasis Rev. 25:315–322. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Spaeth EL, Dembinski JL, Sasser AK, Watson
K, Klopp A, Hall B, Andreeff M and Marini F: Mesenchymal stem cell
transition to tumor-associated fibroblasts contributes to
fibrovascular network expansion and tumor progression. PloS one.
4:e49922009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Galdiero MR, Garlanda C, Jaillon S, Marone
G and Mantovani A: Tumor associated macrophages and neutrophils in
tumor progression. J Cell Physiol. 228:1404–1412. 2013. View Article : Google Scholar
|
|
50
|
Qian B, Deng Y, Im JH, Muschel RJ, Zou Y,
Li J, Lang RA and Pollard JW: A distinct macrophage population
mediates metastatic breast cancer cell extravasation, establishment
and growth. PLoS One. 4:e65622009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Itzkowitz SH and Yio X: Inflammation and
cancer IV. Colorectal cancer in inflammatory bowel disease: The
role of inflammation. Am J Physiol Gastrointest Liver Physiol.
287:G7–G17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gazzaniga S, Bravo AI, Guglielmotti A, van
Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J and Wainstok
R: Targeting tumor-associated macrophages and inhibition of MCP-1
reduce angiogenesis and tumor growth in a human melanoma xenograft.
J Invest Dermatol. 127:2031–2041. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bazzoni F and Beutler B: The tumor
necrosis factor ligand and receptor families. N Engl J Med.
334:1717–1725. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Locksley RM, Killeen N and Lenardo MJ: The
TNF and TNF receptor superfamilies: Integrating mammalian biology.
Cell. 104:487–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou J, Yang Z, Tsuji T, Gong J, Xie J,
Chen C, Li W, Amar S and Luo Z: LITAF and TNFSF15, two downstream
targets of AMPK, exert inhibitory effects on tumor growth.
Oncogene. 30:1892–1900. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pang T, Wang J, Benicky J and Saavedra JM:
Minocycline ameliorates LPS-induced inflammation in human monocytes
by novel mechanisms including LOX-1, Nur77 and LITAF inhibition.
Biochim Biophys Acta. 1820:503–510. 2012. View Article : Google Scholar : PubMed/NCBI
|