Introduction
Heat shock factor 1 (Hsf1) is the predominant
regulator of the heat shock response and has been demonstrated to
be associated with certain tissue specific tumorigenesis (1). Hsf1 protein is upregulated in
malignant tumor tissues of the liver, esophagus, prostate,
lymphatic system, lungs and breasts (2,3).
Inhibition of Hsf1 protein expression suppresses the growth of
certain tumor cell lines and upregulates their sensitivity to
chemotherapeutic agents in vitro (4,5). In
animal models, Hsf1 knockdown inhibits 7,12-dimethylbenz(a)
anthracene-induced skin cancer (6), p53 mutation-induced lymphoma,
n-nitrosodiethylamine-induced hepatocellular carcinoma (HCC)
(2) and epidermal growth factor
receptor II (ErbB2)-associated breast cancer (7). Hsf1 has been associated with multiple
pathways involved in tumorigenesis. For example, Hsf1 participates
in regulating tumor cell protein synthesis, glucose and lipid
metabolism, p53 protein stability (8), chromosome stability, the signal
transduction of ErbB2 (7) and
expression of certain non-heat shock proteins (6,9).
These data support the role of Hsf1 as a potential novel target in
cancer therapy.
Numerous previous studies have indicated that the
Hsf1-mediated heat shock response is critical in modulating cell
transformation resulting from viral oncoproteins, which are
important for tissue specific tumorigenesis, for example human
papillomavirus 16 (HPV16) early genes E6–E7 for cervical carcinoma,
adenovirus early region 1A (E1A) for adenoma of the prostate and
nasal carcinoma and hepatitis B virus-hepatitis B protein (HBV-HBx)
for HCC. For example, HBx activates Hsf1, which is involved in the
upregulation of HBx-induced hepatocyte proliferation (10). Deletion of Hsf1 is able to inhibit
E1A-induced mouse embryonic fibroblast (MEF) cell proliferation
in vitro (11). These
examples demonstrate certain pathways involving Hsf1, however
further studies are required to fully elucidate the association
between Hsf1 and viral oncoproteins in tumorigenesis.
Simian virus 40 (SV40) is a double stranded DNA
virus that is normally expressed in monkey kidney and human brain
tumor and malignant mesothelioma tissue (12). Infection with SV40 leads to animal
tumors (12), however it is
unclear whether SV40 has a similar effect in humans. The proteins
that SV40 encodes, the large T-antigen (TAG) and small t-antigen
(TAG), are strong viral carcinogens and have been widely used to
immortalize normal cells in in vitro tumorigenesis studies
(13). TAG binds to protein
phosphatase 2A (PP2A) and blocks the tumor suppressor activity of
PP2A (14,15). TAG however, is able to transform
host cells by binding to and inactivating the tumor suppressors p53
and phosphorylated retinoblastoma protein (pRb) (16). In addition to its association with
tumor suppressors, SV40/TAG is able to induce the expression of
molecular chaperones such as heat shock protein 70 (Hsp70) and
binding immunoglobulin protein, which in turn promote the cell
transformation activity of SV40/TAG (16,17).
Hsf1 is a unique transcription factor of Hsp70. This suggests that
the Hsf1-mediated heat shock response may be important for
SV40/TAG-induced cell transformation.
The aim of the current study was to investigate the
roles of Hsf1 in the tumorigenesis of SV40/TAG-transformed MEF
cells, by comparing the effects of Hsf1 knockout MEF cells
(MEF/Hsf1-/-), MEF/Hsf1-/- expressing mouse Hsf1 cDNA (MEf/mHsf1)
and wild type (wt) MEF cells. The tumor formation and metastatic
capabilities of SV40/TAG-transformed MEF cells was investigated in
athymic nude mice. The protein expression levels of the
angiogenesis markers; cluster of differentiation 34 (CD34),
vascular endothelial growth factor (VEGF) and factor VIII related
antigen (FVIII/Rag) were investigated immunohistochemically in the
resulting tumor tissues. Using western blotting, the expression
levels of p53 and pRb were measured, in addition to a range of heat
shock proteins. Coimmunoprecipitation was used to investigate
proteins which associate with SV40/TAG.
Materials and methods
Cell lines and plasmids
MEF/wt and MEF/Hsf1-/- cells were generated from
E12.5 embryos from a C57B16/V129 background (donated by Dr
Xianzhong Xiao from the Central South University School of
Medicine, Changsha, China). The cells were transiently transfected
with pcDNA-SV40/TAG (Addgene, Cambridge MA 02139 USA) and
immortalized by passaging the cells for a maximum of 30
generations. To generate the MEF/mHsf1 cell line, the retroviral
packaging cell line HEK293-ampho cells (American Type Culture
Collection, Mansassas, VA, USA) were transiently transfected with
the recombinant retrovirus vector 4 g pWZL-Blas-ticitin-mFlag-Hsf1.
Following a 24-h transfection, the supernatants were collected by
centrifugation at 960 × g for 10 min and mixed with 2 µg/ml
polybrene (Sigma-Aldrich, St. Louis, MO, USA) and used to infect
the MEF/Hsf1-/- cell lines, generating the MEF/mHsf1 cell line.
Athymic nude mice subcutaneous
engraftment assay
Thirty male Balb c-nu/nu specific pathogen free
athymic nude mice, (4 weeks old; body weight 16 g) were purchased
from the Experimental Animal Research Institute of the Chinese
Academy of Medical Sciences (Beijing, China). Mice were housed in a
sterile room and sacrificed by placing in a sealed box containing
CO2. The protocol approved by the Animal Core Facility
of the Experimental Animal Research Institute of the Chinese
Academy of Medical Sciences (no. HUSOM 2015-036). Each mouse was
anesthetized by injection of 0.1 ml/10 g body weight FFm-mix
(Fentanyl citrate, Fluanisone and Midazolam). For the athymic nude
mice engraftment assay, 5×105 cells were subcutaneously
injected into the craniodorsal area of the right leg. Following
engraftment, the mice were continuously fed for a maximum of 50
days. The time taken for tumor formation was recorded, and the
tumor volume was measured using the formula a(b)2/2 (where a
represents the longest tumor diameter and b represents the shortest
tumor diameter) (18).
MTT and colony formation assays
For the MTT assay, 103 cells were seeded
in 96-well plates and cultured for 24, 48, 72 and 96 h. The cells
were incubated with Dulbecco's modified Eagle's medium (DMEM;
Thermo Fisher Scientific, Grand Island, NY, USA) containing
20µM MTT (Life Technologies, Grand Island, NY, USA) for 4 h
and terminated by adding 0.1% NP-40 (0.1 ml)/isopropanol lysis
buffer (100 ml) for 10 min. The absorbance was measured at a
wavelength of 590 nm using Tecan Infinite F500 (Tecan Männedorf,
Switzerland). For the colony formation assay, 103 cells
were seeded into the 6-well plates and cultured for 7 days. The
cells were washed three times with phosphate-buffered saline (PBS)
and stained with 1% crystal violet (Sigma-Aldrich) for 20 min. The
number of colonies was then counted manually.
Cell cycle analysis
The cells were synchronized by serum starvation and
seeded into 6-well plates at a concentration of 5×105,
then were cultured for 24 h in complete media (DMEM with 1X
penicillin-streptomycin, 10 mM glutamine and 10% fetal bovine
serum; all from Thermo Fisher Scientific). The cells were then
trypsinized (Invitrogen Life Technologies, Grand Island, NY, USA)
and fixed in 70% precooled alcohol overnight at 4°C. The cells were
washed in PBS twice to remove the ethanol and resuspended in 500
µl pyridine iodide solution (containing 50 µg/ml
RNase-A; Invitrogen Life Technologies) in the dark for 30 min at
room temperature. The cell cycle was then measured using flow
cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA)
Immunohistochemistry
Tumor tissue was fixed in 4% paraformaldehyde/PBS
for 1 week, embedded in paraffin and sliced into 4 µm tissue
slices by a microtome (Leica RM 2235, Wetzlar, Germany).
Paraffin-embedded tumor tissues were deparaffinized and quenched in
3% H2O2 (Sigma-Aldrich) to remove the
endogenous peroxidase activity. Following antigen retrieval in 0.01
M sodium folic acid buffer (Sigma-Aldrich), the slides were blocked
in 10% normal rabbit serum (Santa Cruz Biotechnology, Inc., Dallas
TX, USA) for 30 min. Subsequently, the slides were incubated with
the following primary antibodies: Mouse monoclonal anti-VEGF (Santa
Cruz Biotechnology, Inc.; cat no. SC-7269; dilution 1:100), mouse
monoclonal anti-CD34 antibody (Santa Cruz Biotechnology, Inc., cat.
no. SC-7324; dilution 1:100), mouse monoclonal anti-FVIII/Rag
antibody (America Diognostica USA, cat. no. ESvWF-10; dilution
1:100), overnight at 4°C. Slides were then incubated with
HRP-conjugated anti-rabbit IgG and HRP-conjugated anti-mouse IgG
secondary antibody (Santa Cruz Biotechnology, Inc., dilution
1:200). The signal was developed with diaminobenzidine
(Sigma-Aldrich) and the slides were counterstained with hematoxylin
and eosin (Sigma-Aldrich). The expression of VEGF, CD34 and
FVIII/Rag were quantified as previously published (19). Student's t-test was used for
statistical analysis. For measurement of the VEGF expression in the
tumor tissues, the brown positive signals were measured by Image
Pro-Plus software. The integral optical density of 10 fields was
measured and averaged. The immunohistochemistry staining signal
density of CD34 and FVIII/Rag were measured in 10 randomly selected
fields. The average of 10 views was used to represent the new
vessel grown in the tumor tissues.
Western blotting and
coimmunoprecipitation
The cells were lysed in NP-lysis buffer (50 mM
Tris-HCl, pH 7.4; 150 mM NaCl, 1% NP-40 and 1X cocktail protease
inhibitor (Sigma-Aldrich). The protein concentration was measured
using a bicinchoninic acid assay kit (Thermo Fisher Scientific).
Equal amounts of protein were separated by 10% SDS-PAGE (Invitrogen
Life Technologies) and transferred onto nitrocellulose membranes
(Invitrogen Life Technologies). The membranes were blocked with 5%
non-fat dried milk/Tris-buffered saline-Tween 20 and incubated with
the primary antibodies at 4°C overnight. The antibodies used were
as follows: Rabbit polyclonal anti-Hsf1 antibody (Cell signaling;
cat. no. 4356; working dilution 1:1,000), rabbit anti-Hsp25
antibody (Sigma-Aldrich; cat. no. H0148; working dilution 1:2,000),
mouse anti-Hsp70 antibody (Enzo Life Sciences, Inc., Farmingdale,
NY, USA; cat. no. ADI-SPA-810; working dilution 1:1,000), mouse
anti-heat shock cognate protein 70 (Hsc70) antibody (Enzo Life
Sciences, Inc.; cat. no. ADI-SPA-820; working dilution 1:1,000),
mouse anti-Hsp90 α antibody (Enzo Life Sciences, Inc.; cat. no.
ADI-SPA-835; working dilution 1:1,000). Membranes were subsequently
incubated with horseradish peroxidase-conjugated secondary
antibodies for 1 h at room temperature. The membranes were
developed using enhanced chemiluminescence and exposed to X-ray
film (Thermo Fisher Scientific). Protein quantification was
performed with Quantity One 4.6 software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). For immunoprecipitation, the assay was
performed as previously published (20). Briefly, 1 mg protein was
pre-cleaned with 30 µl protein-A agarose beads and then
incubated with 2 µg antibody at 4°C overnight. Subsequently,
the sample was incubated with protein A agarose beads (Invitrogen
Life Technologies) for 2 h. The immunoprecipitated protein
complexes were subjected to immunoblotting with rabbit
anti-SV40/TAG (cat. no. V-300), rabbit anti-p53 (cat. no. sc-6243)
and pRB (cat. no. sc-7905) antibodies (Santa Cruz Biotechnology,
Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
software, version 13.0 (SPSS, Inc., Chicago, IL, USA). Student's
t-test was used for paired data that were normally distributed.
Comparisons among values of more than two groups were performed
using analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
Hsf1 promotes SV40-immortalized MEF cell
proliferation by regulating the cell cycle at the G1 and
S phases
In order to determine the role of Hsf1 in the cell
transformation induced by SV40/TAG, three genotypes of SV40/TAG
transformed MEF cell lines were established: MEF/wt, MEF/Hsf1-/-
and MEF/mHsf1 cells (MEF/Hsf1-/- cells expressing mouse Hsf1 cDNA).
The expression of Hsf1 in these three cell lines was investigated
by immunoblotting. As presented in Fig. 1A, Hsf1 in MEF/Hsf1-/- cells. The
proliferation of these three cell lines was investigated using an
MTT and a colony formation assay. MEF/wt and MEF/mHsf1 cells
exhibited a similar rate of proliferation and colony formation, and
were observed to proliferate significantly faster than the
MEF/Hsf1-/- cells (Fig. 1B–D). The
result of the cell cycle analysis indicated that the number of
MEF/wt and MEF/mHsf1 cells at G1 phase were 25 and 22%
respectively, which was significantly increased to 45% in the
MEF/Hsf1-/- cells (P<0.01; Fig.
1E). By contrast, the number of MEF/Hsf1-/- cells at the S and
G2 phases was significantly reduced compared with the
MEF/wt and MEF/mHsf1 cells (P<0.05; Fig. 1E). These results indicate that
knockout of Hsf1 inhibits the proliferation of SV40/TAG transformed
MEF cells in vitro by blocking the cell cycle at the
G1 phase.
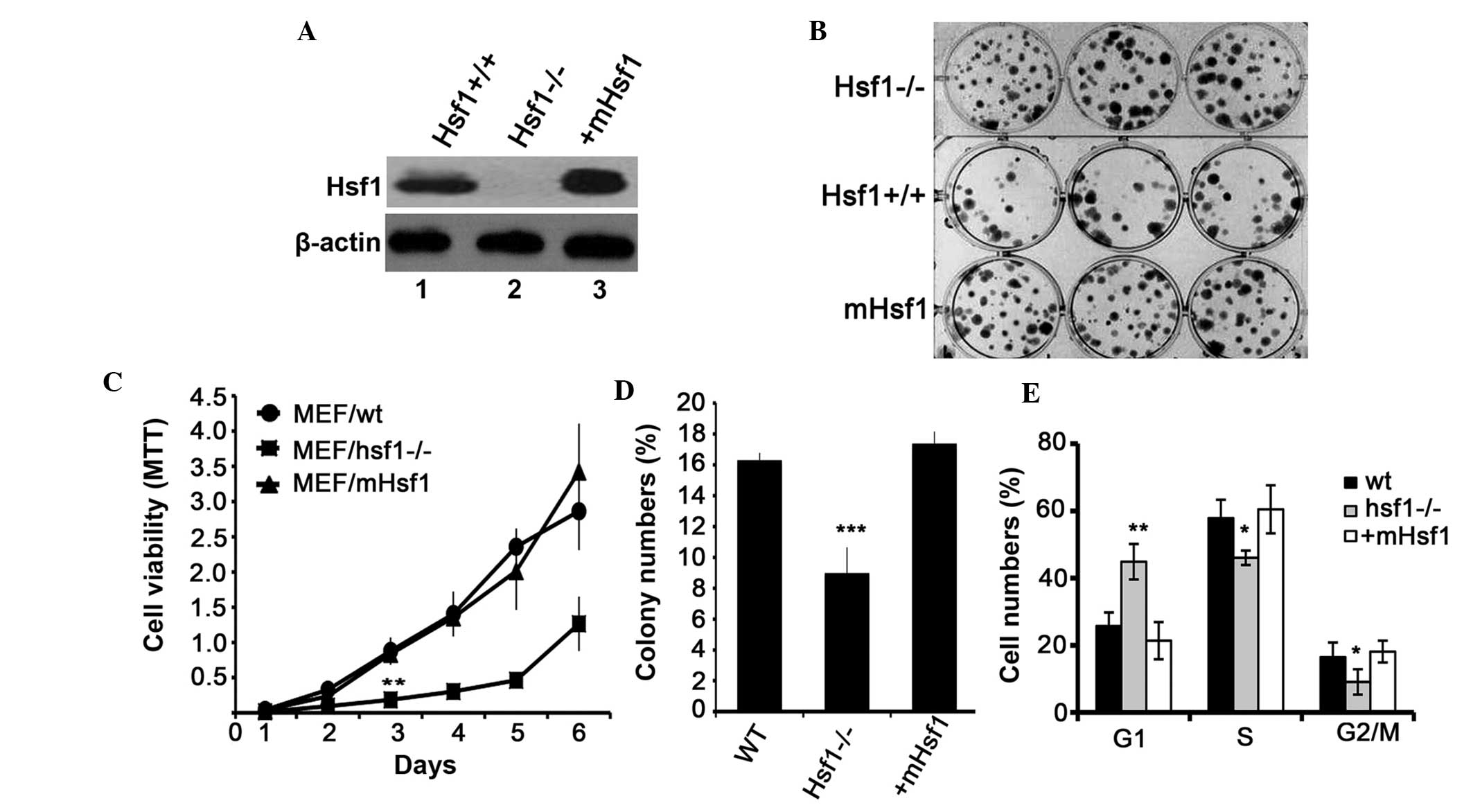 | Figure 1Hsf1 knockout inhibits MEF cell
proliferation. (A) Expression of Hsf1 proteins in the
SV40/TAG-transformed MEF cell lines: Lane 1, MEF/wt; lane 2,
MEF/Hsf1-/-; and lane 3, MEF/mHsf1. (B) Clone formation of the
three MEF cell lines in flat cloning assay. (C) The growth
viability of the three MEF cell lines in an MTT assay. (D) The
quantification of colony-forming efficiency of the three MEF cell
lines by flat cloning assay. (E) The effects of Hsf1 on the cell
cycle of the three MEF cells. *P<0.05,
**P<0.01, ***P<0.001, MEF/Hsf1-/- cells
vs. MEF/wt and MEF/mHsf1 cells. Hsf1, heat shock factor 1; MEF,
mouse embryonic fibroblast; SV40/TAG, simian virus 40/T antigen;
wt, wild type; mHsf1, Hsf1 null MEF cells expressing mouse
Hsf1. |
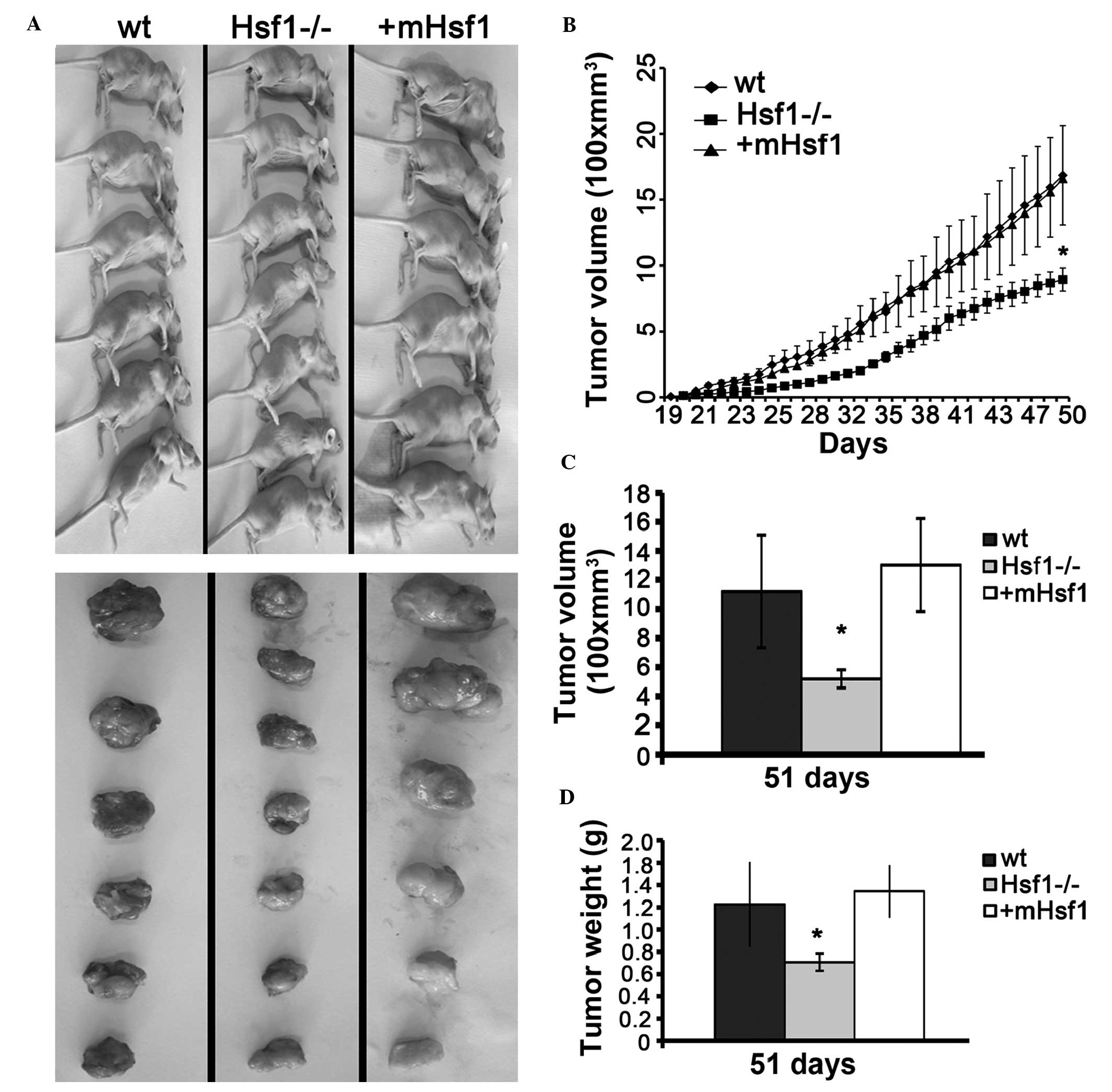
Knockout of Hsf1 inhibits the growth of
fibroblastomas derived from MEF cell lines in athymic nude
mice
SV40/TAG is able to completely transform cells into
malignant tumor cells (16). To
determine the roles of Hsf1 in the SV40/TAG-mediated malignant
transformation of MEF cells, MEF/wt, MEF/Hsf1-/- and MEF/mHsf1
cells were engrafted subcutaneously into athymic nude mice and the
tumor formation and growth were measured. As presented in Fig. 2A, tumors had formed in all athymic
nude mice by day 29 following engraftment. There was no significant
difference in the time taken for tumor formation between the mice
engrafted with MEF/wt, MEF/Hsf1-/- and MEF/mHsf1 cells. However,
the growth of the MEF/wt and MEF/mHsf1 tumors was significantly
faster compared with that of the MEF/Hsf1-/- tumors (P<0.05;
Fig. 2B). No difference in tumor
growth rate was observed between the MEF/wt and MEF/mHsf1 groups
(Fig. 2A and B). Quantitative
results indicated that the weights and volumes of the MEF/wt and
MEF/mHsf1 tumors were significantly greater compared with the
MEF/Hsf1-/- group (P<0.05; Fig. 2C
and D). No difference in tumor size and weight was observed
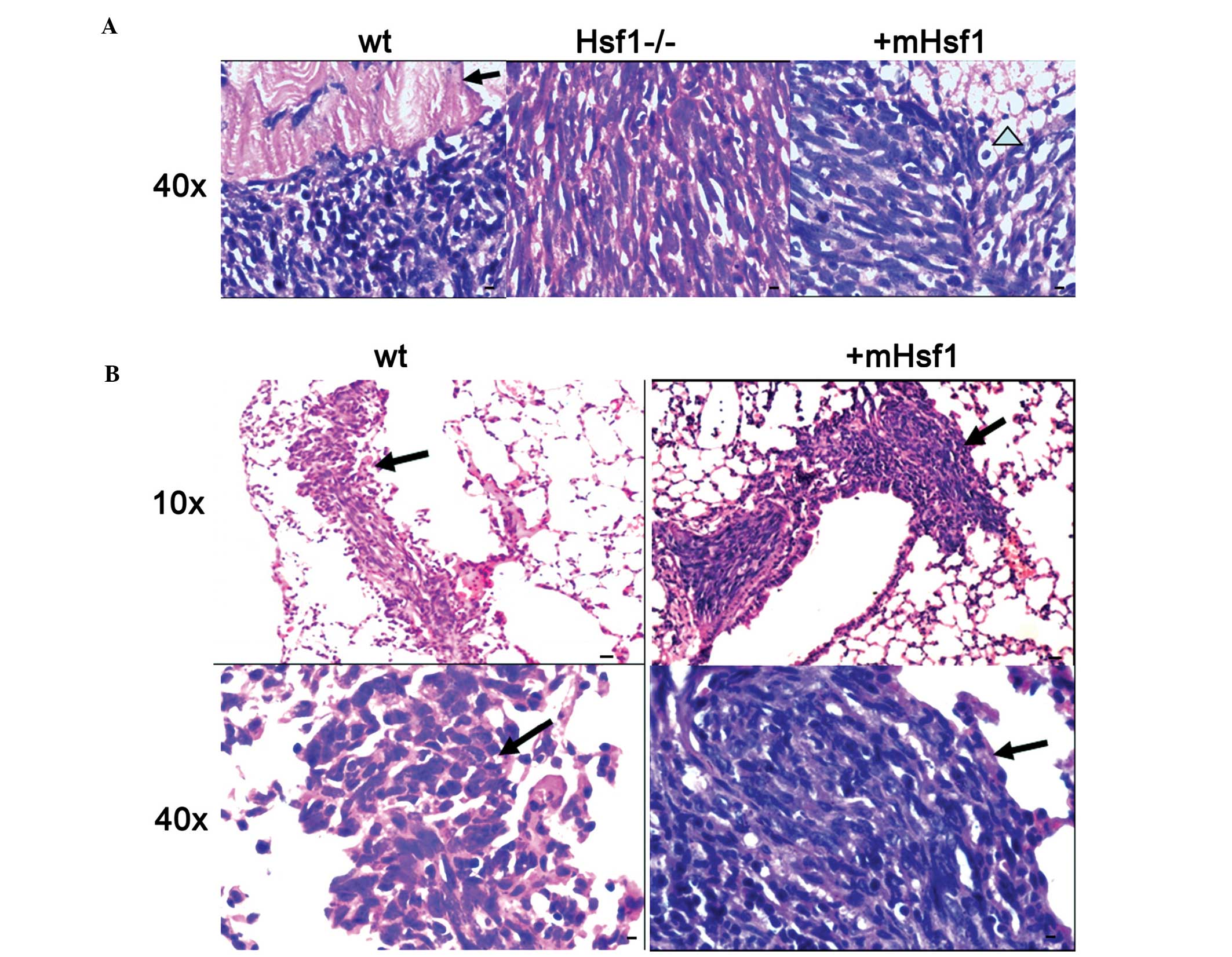
between MEF/wt and MEF/mHsf1 tumors. Histological studies confirmed
that fibro-sarcomas formed from all of the three cell lines
engrafted into athymic nude mice (Fig.
3A). The MEF/wt and MEF/mHsf1 fibrosarcomas exhibited greater
numbers of necrotic foci, and increased levels of pathological
mitosis and peripheral muscle and fat infiltration compared with
that of the MEF/Hsf1-/- fibrosarcoma tissue. These data suggest
that Hsf1 is involved in the regulation of tumor cell proliferation
and development rather than tumor initiation in the SV40/TAG
transformation model.
Knockout of Hsf1 results in
downregulation of tumor angiogenesis
In order to determine the metastatic potential of
these fibrosarcomas, the main organs (brain, liver, lung, spleen
and lymph nodes) of the tumor-bearing mice were screened using
hematoxylin and eosin staining. Histology from 2 of 6 MEF/wt mice
(33.33%) and 1 of 6 MEF/mHsf1 mice (16.67%) exhibited lung
metastasis (Fig. 3B). No
metastasis was observed in the 6 MEF/Hsf1-/- mice. Further studies
using immunohistochemical staining indicated that the expression of
VEGF, CD34 and FVIII/Rag, the three hallmarks of angiogenesis, were
significantly upregulated in the MEF/wt and MEF/mHsf1-derived
fibrosarcoma tissue compared with the MEF/Hsf1-/- fibrosarcoma
tissues (Fig. 4A and B). These
data are consistent with previous reports (7) and suggest that Hsf1 is involved in
the regulation of tumor metastasis in SV40/TAG-induced
fibrosarcoma.
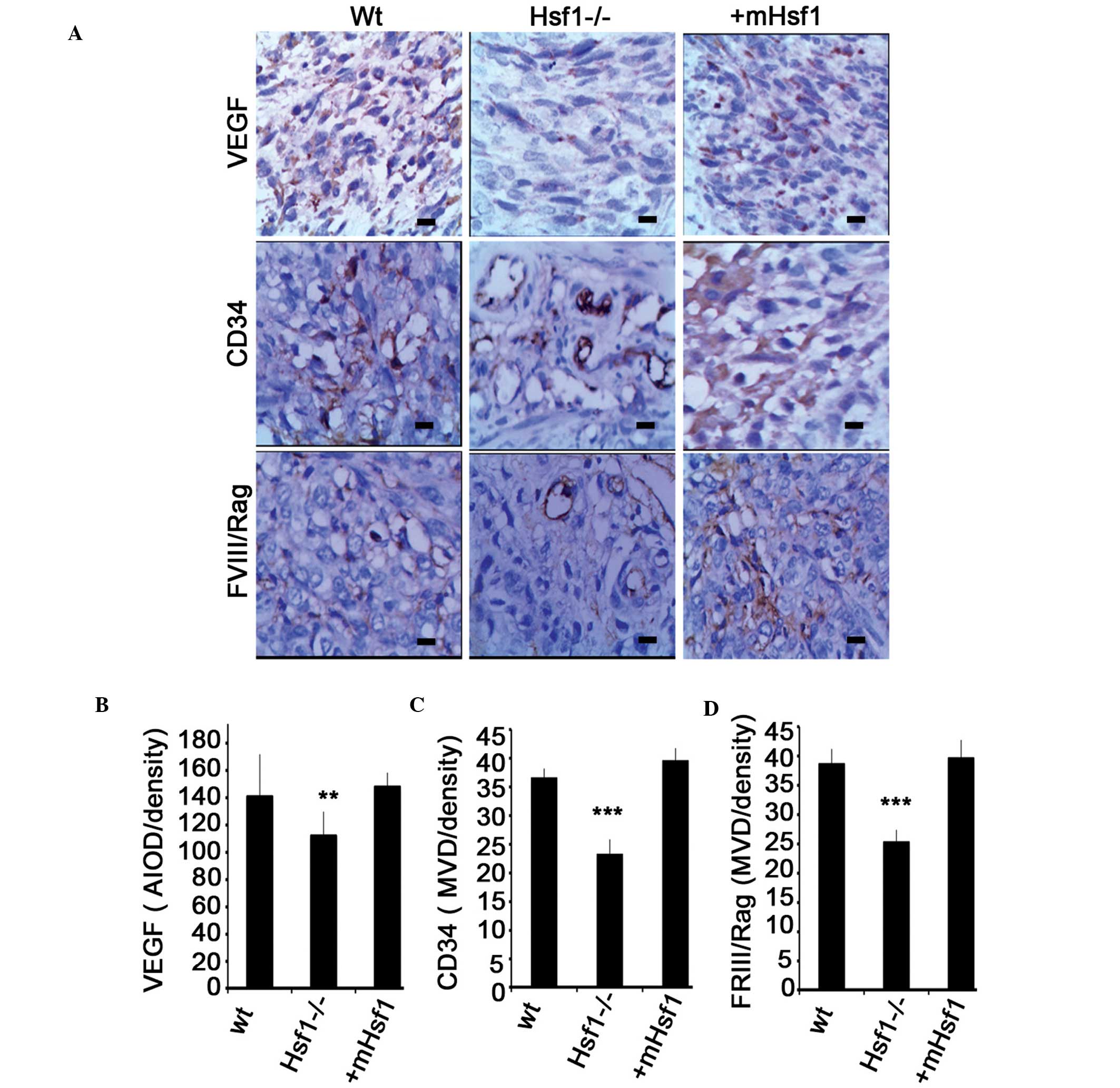 | Figure 4The expression of VEGF, CD34 and
FVIII/Rag proteins in fibrosarcoma. (A) The immunohistochemical
staining of the expression of VEGF, CD34 and FVIII/Rag proteins in
the wt, Hsf1-/- and mHsf1 fibrosarcoma tissues. The images were
taken using a 40X objective. (B) Quantification of the expression
of VEGF in the fibrosarcoma tissue with the AIOD method. The
quantification of the expression of (C) CD34 and (D) FVIII/Rag in
the Hsf1-/- fibrosarcoma using the MVD method. Data are presented
as the mean ± standard deviation, one-way analysis of variance.
**P<0.01, ***P<0.001 vs. wt and mHsf1
cells. VEGF, vascular endothelial growth factor; CD34, cluster of
differentiation 34; FVIII/Rag, factor VIII related antigen; wt,
wild type; Hsf1, heat shock factor 1; mHsf1, Hsf1 null MEF cells
expressing mouse Hsf1. AIOD, average integral optical density. |
Hsf1 is associated with the expression of
p53 and pRb proteins
Cell transformation by SV40/TAG is associated with
p53 and pRb, two tumor suppressors. SV40/TAG binds to p53 and pRb
and suppresses their transcriptional activities, which in turn are
able to induce cell transformation (12). Hsf1 is reported to promote p53
protein degradation by upregulating the expression of the
proteasome subunits proteasome subunit β type-5 and gankynin or
αB-crystallin (8,11). Therefore the deregulation of p53 or
pRb expression may be involved in the growth inhibition of SV40/TAG
transformed MEF/Hsf1-/- cells and the corresponding fibrosarcoma.
Immunoblotting indicated that the protein expression levels of p53,
pRb and their downstream target p21 were significantly upregulated
in MEF/Hsf1-/- cells (Fig. 5A;
lane 2) and the corresponding fibrosarcoma tissue (Fig. 5B; lanes 2, 5 and 8) when compared
with the MEF/wt and MEF/mHsf1 cells (Fig. 5A; lanes 1 and 3) and their
corresponding fibrosarcoma tissues (Fig. 5B; lanes 1, 3, 4, 6, 7 and 9; and
Fig. 5D). Furthermore, the
expression of the SV40/TAG protein, which is similar to p53 and
pRb, was upregulated in fibrosarcoma/Hsf1-/- tissue compared with
its expression in the fibrosarcomas derived from MEF/wt and
MEF/mHsf1 cells (Fig. 5A and B).
To investigate the expression of heat shock proteins underlying
Hsf1 in these tumor tissues, the expression levels of Hsp25, Hsp70
and Hsp90 were measured. The results indicated that the expression
of Hsp25 was downregulated in MEF/Hsf1-/- cells (Fig. 5A; lane 2) and in the corresponding
fibrosarcoma (Fig. 5B; lanes 2, 5
and 8) when compared with the MEF/wt and MEF/mHsf1 cells (Fig. 5A; lanes 1 and 3) and their
corresponding fibrosarcomas (Fig.
5B). No difference in the expression levels of Hsp70, heat
shock cognate protein 70 (Hsc70) and Hsp90 was observed in the
three cell lines and their corresponding fibrosarcomas (Fig. 5A and B). Immunoblotting of β-actin
indicated that protein loading was equal. Fig. 5C and D represent the quantification
of the expression of the corresponding proteins in Fig. 5A and B. Taken together, these data
indicate that knockout of Hsf1 results in the upregulation of p53
and additionally in the upregulation of pRb, p21 and SV40/TAG
proteins. Upregulation of SV40/TAG does not inhibit p53
transcriptional activity in MEF/Hsf1-/- and the derived
fibrosarcoma tissues.
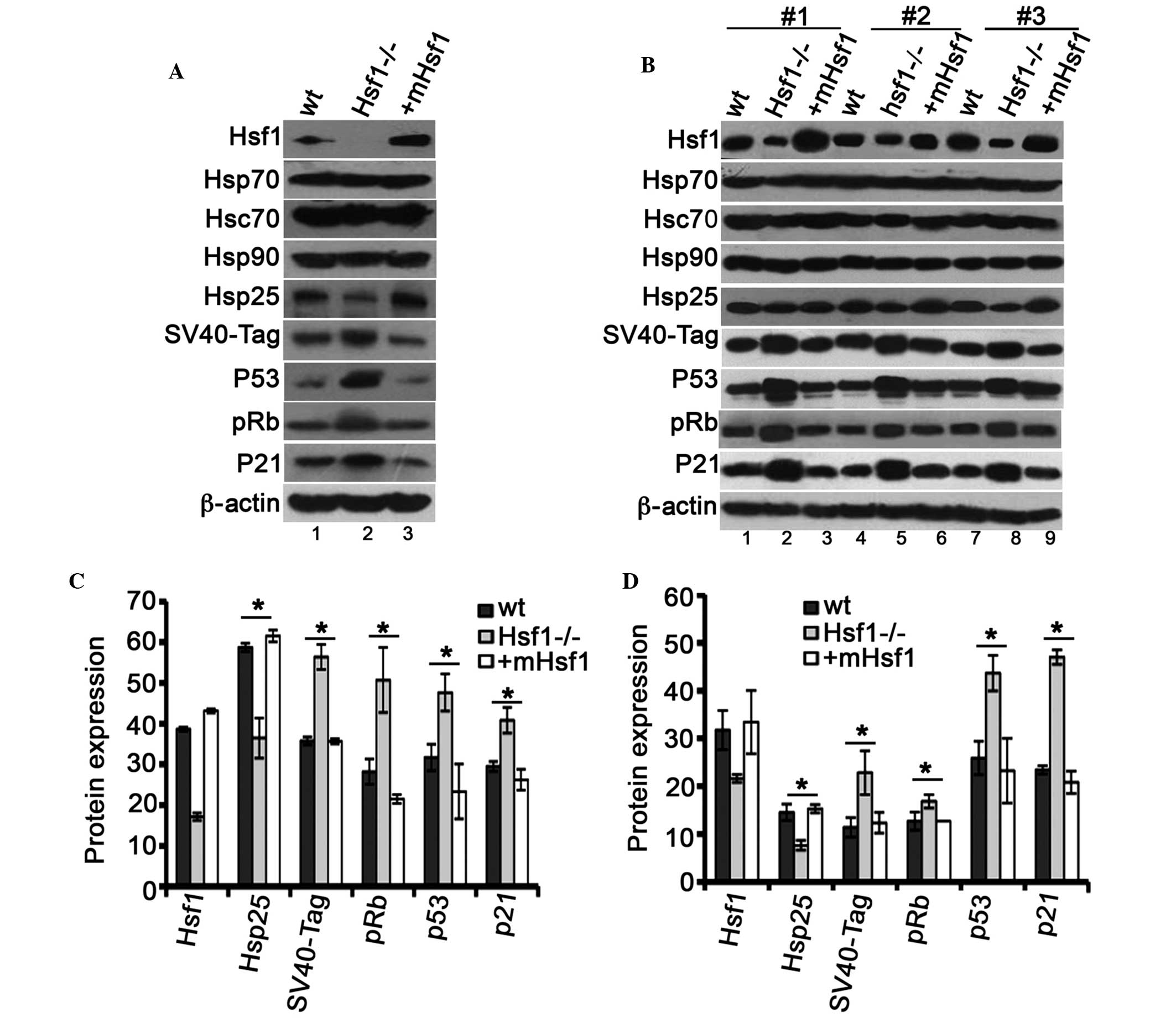 | Figure 5Hsf1 is associated with the
expression of p53 and pRb proteins. (A) The expression of Hsf1,
Hsp25, Hsc70, Hsp70, Hsp90, SV40/TAG, p53, p21 and pRb proteins
were immunoblotted in MEF/wt (lane 1), MEF/hsf1-/- (lane 2) and
MEF/mHsf1 (lane 3) cells. (B) The immunoblotting of the above
proteins in the corresponding fibrosarcoma tissues. Experiments
were repeated 3 times (#1–3). The quantification of the expression
of Hsf1, Hsp25, SV40/TAG, p53, pRB and p21 in the three (C) MEF
cell lines and their corresponding (D) fibrosarcomas.
(*P<0.05). hsf1, heat shock factor 1; pRb,
phosphorylated retinoblastoma protein; Hsp25, heat shock protein
25; Hsc70, heat shock cognate protein 70; SV40/TAG, simian virus
40/T antigen; MEF, mouse embryonic fibroblast; wt, wild type;
mHsf1, Hsf1 null MEF cells expressing mouse Hsf1. |
Knockout of Hsf1 reduces the interaction
between SV40/TAG and p53 and pRb
The immunoblotting data indicates that knockout of
Hsf1 inhibits the suppression of p53 transcriptional activity by
SV40/TAG, which suggests that Hsf1 is involved in the association
between p53 and SV40/TAG. To investigate this, the interaction
between SV40/TAG and p53/pRb was measured in the three types of MEF
cells using immunoprecipitation assays. The amount of p53 and pRb
proteins that were coimmunoprecipitated with SV40/TAG in the
MEF/Hsf1-/- cells was reduced compared with that in the MEF/wt and
MEF/mHsf1 cells. No interaction between SV40/TAG and Hsc70 was
observed in the three MEF cell lines (data not shown). The
expression of endogenous p53, pRb, β-actin and ectopic SV40/TAG in
the cell lysates that were applied for the immunoprecipitation
assay are presented in Fig. 6B.
These data indicate that the knockout of Hsf1 reduces the
association of SV40/TAG with p53 and/or pRb.
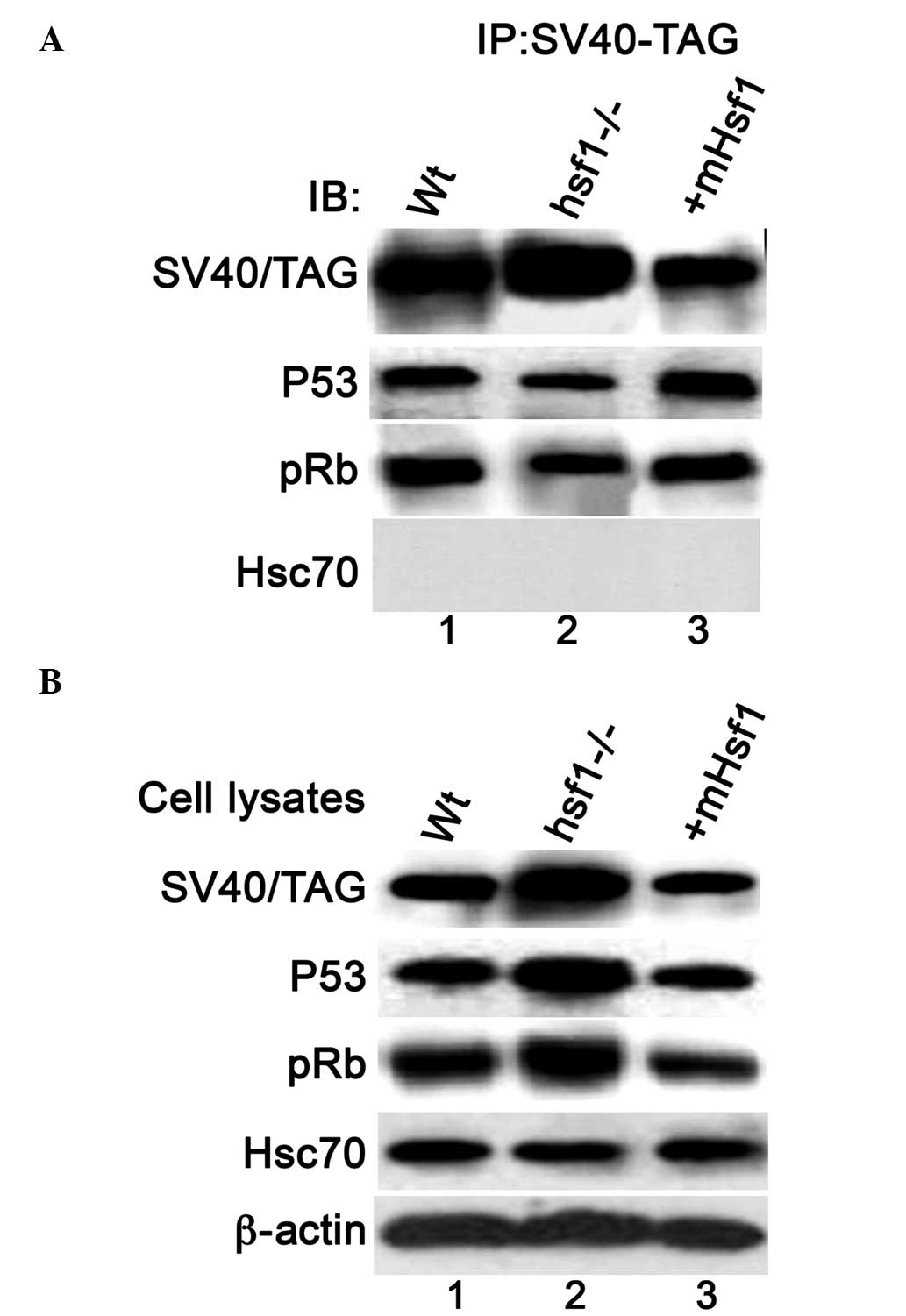 | Figure 6The interaction of SV40/TAG with p53,
pRb and Hsc70 in the three MEF cell lines. (A)
Coimmunoprecipitation of p53, pRb and Hsc70 with SV40/TAG. SV40/TAG
proteins were immunoprecipitated from the cell lysates of the
MEF/wt (lane 1), MEF/Hsf1-/- (lane 2) and MEF/mHsf1 (lane 3) cells.
The immunoprecipitated protein complexes were immunoblotted with
antibodies against p53, pRB and Hsc70. (B) The immunoblotting of
the expression of SV40/TAG, p53, pRb and Hsc70 proteins in the cell
lysates that were subjected to coimmunoprecipitation. SV40/TAG,
simian virus 40/T antigen; pRb, phosphorylated retinoblastoma
protein; Hsc70, heat shock cognate protein 70; MEF, mouse embryonic
fibroblast; wt, wild type; Hsf1, heat shock factor 1; mHsf1, Hsf1
null MEF cells expressing mouse Hsf1; IP, immunoprecipitate; IB;
immunoblot. |
Discussion
Hsf1 has been demonstrated to be associated with
tumorigenesis in animal models and in humans (1). It has been reported that Hsf1 is
important for the oncogenic processes mediated by Ras, p53 and E1A
(6,11). In the current study, evidence that
Hsf1 is additionally involved in SV40/TAG-induced cell
transformation is presented. MEF/wt and MEF/Hsf1-/- cells were
immortalized by SV40/TAG and formed fibrosarcomas following
engraftment subcutaneously in athymic nude mice (Fig. 2). However, knockout of Hsf1 blocked
the cell cycle at the G1 phase and reduced the number of
cells in the S and G2 phases. This resulted in the
inhibition of MEF/Hsf1-/- cell proliferation in vitro and of
MEF/Hsf1-/-fibrosarcoma growth in vivo in the athymic nude
mice (Figs. 1 and 2). Further investigation suggested that
the knockout of Hsf1 is able to upregulate the protein expression
of the tumor suppressors p53, pRb and p21 in addition to SV40/TAG
(Fig. 5). However, the
associations between SV40/TAG and p53 and pRb were significantly
reduced in MEF/Hsf1-/- cells, which may provide an explanation for
the slow growth of the Hsf1-/- fibrosarcoma. Furthermore, the data
suggested that the knockout of Hsf1 suppressed lung metastasis of
the fibrosarcoma by reducing angiogenesis (Fig. 3). Taken together, these data
provide further evidence to support the fact that Hsf1 is an
important factor for the growth and metastasis of viral
oncogene-induced tumors.
There is substantial evidence in support of the
involvement of Hsf1 in tumor initiation and the development of
tumors induced by p53-mutation, H-Ras and ErbB2-mutations in mouse
models (7,9). However, its roles in the
tumorigenesis of viral oncogene induced tumors remain unclear. Jin
et al (11) reported that
MEF/wt and MEF/Hsf1-/- cells were immortalized by E1A, however the
proliferation and colony formation of the E1A-immortalized
MEF/Hsf1-/- cells was slower compared with the MEF/wt cells.
Upregulation of p53 protein through the reduced expression of
αB-crystallin expression in MEF/Hsf1-/- cells was suggested to
explain this slow proliferation (11). αB-crystallin has been reported to
form a complex with F-box only protein 7 and p53, mediating p53
degradation through the proteasomal pathway (11). However, there are inadequate in
vivo studies to suggest whether the E1A-immortalized MEF cells
are malignant. In the current study, SV40/TAG transformed
MEF/Hsf1-/- and MEF/wt cells in vitro, and the transformed
cells formed fibrosarcomas when subcutaneously engrafted into
athymic nude mice. However, the growth of the MEF/Hsf1-/- cells and
their corresponding fibrosarcomas was significantly slower compared
with MEF/wt cells. These data indicate that Hsf1 is important in
maintaining viral tumor cell proliferation rather than initiating
transformation.
SV40/TAG induces cell transformation by targeting
two tumor suppressors, p53 and pRb. SV40/TAG interacts with and
inhibits the transcriptional activities of p53 and pRb, which
result in the deregulation of the cell cycle and cell
transformation (13,21). The results of the current study
indicate that the expression of p53 and pRb are upregulated in
MEF/Hsf1-/- cells, which are consistent with the previously
reported results, in which Hsf1 knockout resulted in p53 protein
stabilization by E1A (11).
Although it is unclear which mechanism results in the accumulation
of p53 and pRb proteins in SV40/TAG-transformed MEF/Hsf1-/- cells,
the current study indicated that in addition SV40/TAG protein was
upregulated in the MEF/Hsf1-/- cells and fibrosarcoma, and the
upregulation of SV40/TAG did not inhibit the transcriptional
regulation of p21 expression by p53. This suggests that the
association between SV40/TAG and p53 or pRb may be blocked in
MEF/Hsf1-/- cells and fibrosarcoma tissue. The
coimmunoprecipitation data demonstrated that the interaction
between SV40/TAG and p53 or pRb is significantly reduced in
MEF/Hsf1-/- cells compared with that in MEF/wt and MEF/mHsf1 cells,
despite the SV40/TAG-induced upregulation of p53 and pRb proteins
in MEF/Hsf1-/- cells. It remains unclear which mechanism is
responsible for the reduced interaction between SV40/TAG and p53 or
pRb in the MEF/Hsf1-/- cells. Xi et al (7) reported that knockdown of Hsf1 is able
to reduce the interaction between Raf and Hsp90, which then in turn
reduces the signal transduction from ErbB2 to the mitogen activated
protein kinase pathway, and the ErbB2-mutation-induced breast
cancer occurrence in mouse models. The current study measured the
expression of additional heat shock proteins (Hsp90, Hsp70 and
Hsp25) and observed that Hsp25 alone is significantly reduced in
MEF/Hsf1-/- cells and the corresponding fibrosarcoma, suggesting
that Hsp25 is the predominant target of Hsf1 in
SV40/TAG-transformed MEF cells and their corresponding
fibrosarcomas. A previous study reported that heat shock proteins
are upregulated in SV40 transformed cells (22), suggesting that heat shock proteins
in general serve a role in SV40/TAG transformation (e.g.
anti-apoptosis and cell cycle proliferation). However, whether
Hsp25 is specifically involved in regulating the association
between SV40/TAG and p53 or pRb requires further investigation.
Invasion and metastasis are two important hallmarks
of malignant tumors, and a number of factors have been demonstrated
to be involved in these processes [e.g. VEGF, hypoxia-inducible
factor 1 α(Hif1α) and matrix metalloproteinases]. It has been
reported that Hsf1 is involved in the regulation of tumor
metastasis in breast cancer (7,9).
Upregulation of Hsf1 is associated with worsened prognosis in
breast cancer, HCC and other types of tumor (9). Xi et al (7) reported that knockdown of Hsf1
inhibits breast cancer development and transforming growth
factor-β1 induced epithelial-mesenchymal transition. Gabai et
al (23) reported that Hsf1
regulates breast cancer progression by regulating the expression of
Hif1α and RNA regulator Human antigen R. Mendillo et al
(9) demonstrated that in addition
to regulating heat shock proteins, Hsf1 regulates the expression of
genes that regulate breast cancer cell proliferation, migration and
invasion (9). In the current
study, the fibrosarcomas derived from MEF/wt and MEF/mHsf1 cells
were observed to metastasize to the lungs, however, the
fibrosarcoma derived from MEF/Hsf1-/- did not. Further analysis
indicated that knockout of Hsf1 reduced the angiogenesis of the
fibrosarcoma. This is reflected by the low expression of the
angiogenesis markers including VEGF, CD34 and FVIII/Rag in the
MEF/Hsf1-/- fibrosarcoma compared with the fibrosarcomas that were
derived from MEF/wt and MEF/mHsf1 cells. Tumor analysis
demonstrated that MEF/wt and MEF/mHsf1 fibrosarcomas grew faster,
contained larger mitotic cells and more fat, and exhibited
increased muscle invasion when compared with the MEF/Hsf1-/-
fibrosarcoma. This suggests that Hsf1 is an important factor in
regulating tumor malignancy and metastasis. However, the mechanism
behind this regulation remains unclear. Hsp25 has been reported to
be associated with cancer metastasis (24,25)
and has been targeted as a diagnostic marker for a number of tumors
(26). In addition, Hsp25 was
demonstrated to regulate the stability of a number of transcription
factors such as GATA-1 and Snail (27,28),
which are important in promoting tumor cell growth and metastasis.
The current study indicates that Hsp25 is downregulated in the
MEF/Hsf1-/- cells and fibrosarcoma tissue. However, whether the
downregulation of Hsp25 is involved in regulating the dissociation
of SV40-TAG from p53 and pRb, and the reduced metastatic potential
of the MEF/Hsf1-/- fibrosarcoma requires further investigation.
Taken together, these data provide further evidence to support the
fact that Hsf1 is an important regulator in tumor metastasis.
Using the SV40/TAG immortalized MEF cell model, the
current study demonstrated that Hsf1 is involved in the regulation
of viral-oncogene induced tumor growth and metastasis rather than
tumor initiation. These data provide further evidence to suggest
that Hsf1 may be a potential therapeutic target for
viral-oncogene-induced tumors.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant nos. 30971508 and
81270985).
References
|
1
|
Calderwood SK: Elevated levels of HSF1
indicate a poor prognosis in breast cancer. Future Oncol.
8:399–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin X, Moskophidis D and Mivechi NF: Heat
shock transcription factor 1 is a key determinant of HCC
development by regulating hepatic steatosis and metabolic syndrome.
Cell Metab. 14:91–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Santagata S, Hu R, Lin NU, Mendillo ML,
Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM,
Lindquist S and Ince TA: High levels of nuclear heat-shock factor 1
(HSF1) are associated with poor prognosis in breast cancer. Proc
Natl Acad Sci USA. 108:18378–18383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Whitesell L and Lindquist S: Inhibiting
the transcription factor HSF1 as an anticancer strategy. Expert
Opin Ther Targets. 13:469–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Chen J, Loo A, Jaeger S,
Bagdasarian L, Yu J, Chung F, Korn J, Ruddy D, Guo R, et al:
Targeting HSF1 sensitizes cancer cells to HSP90 inhibition.
Oncotarget. 4:816–829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai C, Whitesell L, Rogers AB and
Lindquist S: Heat shock factor 1 is a powerful multifaceted
modifier of carcinogenesis. Cell. 130:1005–1018. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xi C, Hu Y, Buckhaults P, Moskophidis D
and Mivechi NF: Heat shock factor Hsf1 cooperates with ErbB2
(Her2/Neu) protein to promote mammary tumorigenesis and metastasis.
J Biol Chem. 287:35646–35657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lecomte S, Desmots F, Le Masson F, Le Goff
P, Michel D, Christians ES and Le Dréan Y: Roles of heat shock
factor 1 and 2 in response to proteasome inhibition: Consequence on
p53 stability. Oncogene. 29:4216–4224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mendillo ML, Santagata S, Koeva M, Bell
GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L and Lindquist
S: HSF1 drives a transcriptional program distinct from heat shock
to support highly malignant human cancers. Cell. 150:549–562. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu R, Zhang X, Zhang W, Fang Y, Zheng S
and Yu XF: Association of human APOBEC3 cytidine deaminases with
the generation of hepatitis virus B x antigen mutants and
hepatocellular carcinoma. Hepatology. 46:1810–1820. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin X, Moskophidis D, Hu Y, Phillips A and
Mivechi NF: Heat shock factor 1 deficiency via its downstream
target gene alphaB-crystallin (Hspb5) impairs p53 degradation. J
Cell Biochem. 107:504–515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Colvin EK, Weir C, Ikin RJ and Hudson AL:
SV40 TAg mouse models of cancer. Semin Cell Dev Biol. 27:61–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ludlow JW, Shon J, Pipas JM, Livingston DM
and DeCaprio JA: The retinoblastoma susceptibility gene product
undergoes cell cycle-dependent dephosphorylation and binding to and
release from SV40 large T. Cell. 60:387–396. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pallas DC, Shahrik LK, Martin BL, Jaspers
S, Miller TB, Brautigan DL and Roberts TM: Polyoma small and middle
T antigens and SV40 small t antigen form stable complexes with
protein phosphatase 2A. Cell. 60:167–176. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen W, Possemato R, Campbell KT, Plattner
CA, Pallas DC and Hahn WC: Identification of specific PP2A
complexes involved in human cell transformation. Cancer Cell.
5:127–136. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahuja D, Sáenz-Robles MT and Pipas JM:
SV40 large T antigen targets multiple cellular pathways to elicit
cellular transformation. Oncogene. 24:7729–7745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang J and DeFranco DB: Differential roles
of heat shock protein 70 in the in vitro nuclear import of
glucocorticoid receptor and simian virus 40 large tumor antigen.
Mol Cell Biol. 14:5088–5098. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wapnir IL, Wartenberg DE and Greco RS:
Three dimensional staging of breast cancer. Breast Cancer Res
Treat. 41:15–19. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kraan MC, Smith MD, Weedon H, Ahern MJ,
Breedveld FC and Tak PP: Measurement of cytokine and adhesion
molecule expression in synovial tissue by digital image analysis.
Ann Rheum Dis. 60:296–298. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu YZ, Zhang J, Li S, Wang C, Chu L, Zhang
Z, Ma Z, Wang M, Jiang Q, Liu G, et al: The transcription activity
of heat shock factor 4b is regulated by FGF2. Int J Biochem Cell
Biol. 45:317–325. 2013. View Article : Google Scholar
|
|
21
|
Sáenz Robles MT and Pipas JM: T antigen
transgenic mouse models. Semin Cancer Biol. 19:229–235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Omar RA and Lanks KW: Heat shock protein
synthesis and cell survival in clones of normal and simian virus
40-transformed mouse embryo cells. Cancer Res. 44:3976–3982.
1984.PubMed/NCBI
|
|
23
|
Gabai VL, Meng L, Kim G, Mills TA,
Benjamin IJ and Sherman MY: Heat shock transcription factor Hsf1 is
involved in tumor progression via regulation of hypoxia-inducible
factor 1 and RNA-binding protein HuR. Mol Cell Biol. 32:929–940.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shiota M, Bishop JL, Nip KM, Zardan A,
Takeuchi A, Cordonnier T, Beraldi E, Bazov J, Fazli L, Chi K, et
al: Hsp27 regulates epithelial mesenchymal transition, metastasis,
and circulating tumor cells in prostate cancer. Cancer Res.
73:3109–3119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pavan S, Musiani D, Torchiaro E, Migliardi
G, Gai M, Di Cunto F, Erriquez J, Olivero M and Di Renzo MF: HSP27
is required for invasion and metastasis triggered by hepatocyte
growth factor. Int J Cancer. 134:1289–1299. 2014. View Article : Google Scholar
|
|
26
|
Mese H, Sasaki A, Nakayama S, Yoshioka N,
Yoshihama Y, Kishimoto K and Matsumura T: Prognostic significance
of heat shock protein 27 (HSP27) in patients with oral squamous
cell carcinoma. Oncol Rep. 9:341–344. 2002.PubMed/NCBI
|
|
27
|
Wettstein G, Bellaye PS, Kolb M, Hammann
A, Crestani B, Soler P, Marchal-Somme J, Hazoume A, Gauldie J,
Gunther A, et al: Inhibition of HSP27 blocks fibrosis development
and EMT features by promoting Snail degradation. FASEB J.
27:1549–1560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
de Thonel A, Vandekerckhove J, Lanneau D,
Selvakumar S, Courtois G, Hazoume A, Brunet M, Maurel S, Hammann A,
Ribeil JA, et al: HSP27 controls GATA-1 protein level during
erythroid cell differentiation. Blood. 116:85–96. 2010. View Article : Google Scholar : PubMed/NCBI
|