Introduction
Sepsis, a type of systemic inflammatory response
syndrome (SIRS), is a complication caused by the response of the
whole body, and injured tissues and organs to an infection
(1). In the last decade, the
incidence rates of sepsis and severe sepsis in the population were
4.37 and 2.70 cases per 1,000 individuals, respectively, in
high-income-countries (including seven countries on four
continents); annually, the global mortality rate is as high as
5,300,000 (2). In China, a survey
revealed that, 10 years ago, the incidence and mortality rates of
sepsis were 8.68 and 48.7%, respectively (3). It is difficult to predict, diagnose
and treat patients with sepsis (4). In addition, there are no effective
drugs or medical interventions for treating this disease.
Therefore, the investigation of target genes and microRNAs (miRNAs)
in sepsis is urgently required.
Proinflammatory cytokines, including interleukin-6
(IL-6) and tumor necrosis factor-α (TNF-α), have been implicated as
key mediators in inflammation associated with sepsis (5). Nrf2, a basic leucine zipper
transcription factor, is a regulatory factor for response to
lipopolysaccharides (LPS) and TNF-α by activating the expression of
nuclear factor-κB (NF-κB) during experimental sepsis (6). PTP1B is a target gene for the
treatment of sepsis via the reduction of cardiovascular
inflammation (7). In addition,
miRNAs are small non-coding RNA molecules of 20–23 nucleotides
present in almost all living things, ranging from viruses to higher
animals (8). miRNAs have various
biological functions and are involved in inflammation, metabolism
and tumor processes through a series of signaling pathways
(9). miRNA (miR)-34a (10), miR-150 (11) and miR-15a (10) are considered to be novel indicators
for the early diagnosis and prognosis of sepsis. However, the role
of target genes and miRNAs in sepsis requires further elucidation.
To clarify the role of key genes and miRNAs in sepsis, the present
study aimed to identify potential genes and miRNAs associated with
sepsis, based on the GSE13205 dataset from the Gene Expression
Omnibus (GEO) database (12).
The present study identified differentially
expressed genes (DEGs) in sepsis, and then analyzed the potential
functions of these DEGs using Gene Ontology (GO) functional and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analyses. Subsequently, a protein-protein interaction (PPI) network
between the DEGs was constructed and GO-associated analysis of the
PPI network module was performed. Finally, an integrated regulatory
network was constructed based on the DEGs, miRNAs and transcription
factors (TFs) for the prediction of key targets in sepsis.
Materials and methods
Microarray data
The gene expression profile of GSE13205 was
downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/), which included
muscle biopsy specimens from 13 patients with sepsis from the
General Intensive Care Unit at Karolinska University Hospital
(Huddinge, Sweden) and eight healthy controls from Ersta Hospital
(Stockholm, Sweden). Muscle biopsy samples were obtained from the
lateral portion of the vastus lateralis muscle (10–20 cm above the
knee). The GSE13205 profile was deposited by Fredriksson et
al according to the platform of GPL570 [HG-U133_Plus_2]
Affymetrix Human Genome U133 Plus 2.0 Array. Their study was
approved by the Ethical Committee of Karolinska Institutet
(Stockholm, Sweden) and received local ethics approval for gene
expression analysis at Heriot-Watt University (Edinburgh, Scotland)
(13). Informed consent was
obtained from all patients or close relatives.
Data preprocessing and identification
of DEGs
The microarray data were normalized using the robust
multi-array average function in the Affy package (version 1.52.0;
http://bioconductor.org/packages/release/bioc/html/affy.html)
of R software (version 3.3.2; https://cran.r-project.org/bin/windows/base/)
(14). Subsequently, the Bayesian
method of the Linear Models for Microarray (LIMMA; version 3.30.3;
http://bioconductor.org/packages/release/bioc/html/limma.html)
package in R was used for identifying the DEGs between the samples
from patients with sepsis and those from the healthy controls
(15). The threshold value of DEGs
was set as |log fold change (FC)| >1 and P<0.05.
GO and KEGG pathway enrichment
analyses
GO analysis (http://geneontology.org/page/go-enrichment-analysis)
has become a predictable method for enrichment analysis to
understand biological process (BP), cellular localization and
molecular function (16). KEGG
(http://www.genome.jp/kegg/) is a
well-known database for the systematic analysis of gene function
and genomic information, including the GENES and PATHWAY databases
(17). The Database for
Annotation, Visualization and Integration Discovery (DAVID; version
6.7; https://david-d.ncifcrf.gov/) is a
public high-throughput functional annotation tool, which provides
functional annotation bioinformatics microarray analysis for
integrating data-mining environments and analyzing gene lists
(18). The DEGs were input into
DAVID online for GO and KEGG enrichment analyses. P<0.05 was
considered to be statistically significant.
Analysis of the PPI network
The PPI network was constructed with all the DEGs
using the Search Tool for the Retrieval of Interacting Genes
(STRING) from a well-known online server (version 10.0; http://www.string-db.org/) (19). PPI links with a combined score
>0.4 were identified for constructing the PPI network. The PPI
network was visualized using Cytoscape software (http://cytoscape.org/), and the network topology was
analyzed using CytoNCA (version 2.1.6; http://apps.cytoscape.org/apps/cytonca) (20). The parameter was set as ‘without
weight’.
GO-associated analysis of the PPI
network module
MCODE in Cytoscape was used to search for modules in
the PPI network (21). The modules
were identified when the degree of cut-off was 2, the node score
cut-off was 0.2, the K-core was 2, and the maximum depth was 100.
GO-associated analysis was performed on the module with the maximum
score using Golorize (version: 1.0.0.beta1) (22). In addition, the top five GO terms
in the module were analyzed.
Construction of the integrated
regulatory network
The integrated regulatory network was constructed
based on the DEGs, miRNAs and TFs. miRNAs associated with DEGs were
identified from the mir2disease database (http://www.mir2disease.org/) and miRWalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/)
(23,24). Finally, the miRNA-target gene
links, which overlapped with DEGs, were considered to be closely
associated with sepsis. TFs were selected from the integrated
transcription factor platform (http://itfp.biosino.org/itfp) (25) and TRANSFAC (http://www.gene-regulation.com/pub/databases.html)
among DEGs (26). The DEG-miRNA-TF
network was constructed and visualized using Cytoscape
software.
Results
DEGs in sepsis
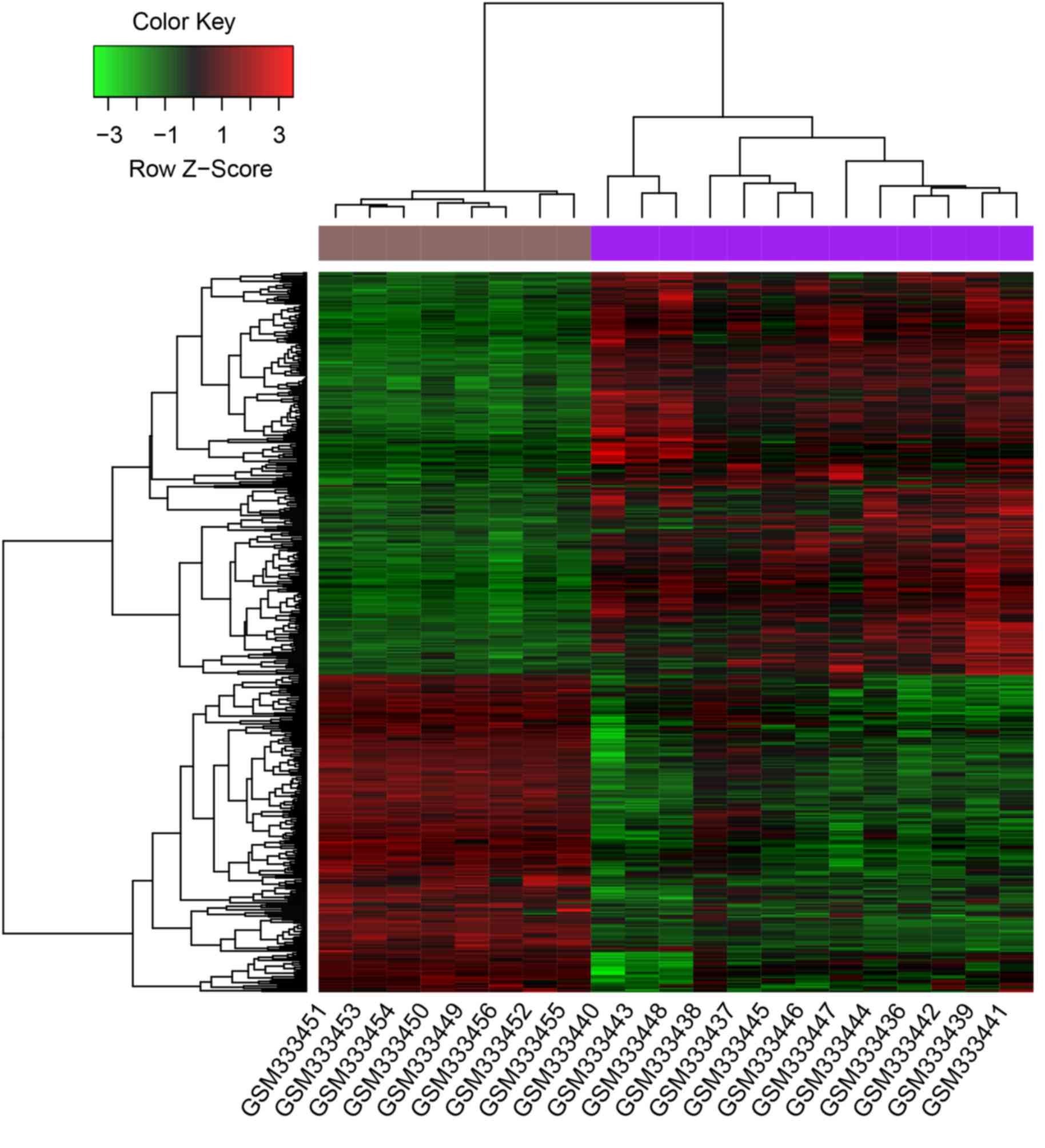
A total of 463 DEGs were identified from patients
with sepsis and healthy controls, which comprised 259 upregulated
DEGs, including MYC and BYSL, and 204 downregulated
DEGs, including MYLK3. The clustering heatmap showed that
DEGs were well distinguished between samples from patients with
sepsis and those from healthy controls (Fig. 1).
Results of GO and KEGG enrichment
analysis
For the upregulated DEGs, all were enriched in 51 GO
BP terms and two KEGG pathways. MAP3K14 was enriched in the
apoptosis pathway (Table I).
Downregulated DEGs were enriched in 37 GO BP terms and five KEGG
pathways, including the adipocytokine and calcium signaling
pathways (Table II).
 | Table I.Results of GO and KEGG enrichment on
upregulated differentially expressed genes. |
Table I.
Results of GO and KEGG enrichment on
upregulated differentially expressed genes.
| Category | Term | Count | P-value |
|---|
| GO term BP | GO:0045768-Positive
regulation of anti-apoptosis | 5 | 6.2E-4 |
| GO term BP |
GO:0045767-Regulation of
anti-apoptosis | 5 | 1.5E-3 |
| GO term BP |
GO:0022613-Ribonucleoprotein complex
biogenesis | 9 | 2.7E-3 |
| GO term BP | GO:0010033-Response
to organic substance | 20 | 3.2E-3 |
| GO term BP |
GO:0006366-Transcription from RNA
polymerase II promoter | 10 | 3.9E-3 |
| KEGG pathway | Hsa04210:
Apoptosis | 5 | 1.3E-2 |
| KEGG pathway | Hsa00480:
Glutathione metabolism | 4 | 1.6E-2 |
| Table II.Results of GO and KEGG enrichment on
downregulated differentially expressed genes. |
Table II.
Results of GO and KEGG enrichment on
downregulated differentially expressed genes.
| Category | Term | Count | P-value |
|---|
| GO term BP | GO:0007517-Muscle
organ development | 14 | 3.68E-8 |
| GO term BP | GO:0006936-Muscle
contraction | 11 | 8.74E-7 |
| GO term BP | GO:0003012-Muscle
system process | 11 | 2.05E-6 |
| GO term BP |
GO:0044057-Regulation of system
process | 13 | 1.60E-5 |
| GO term BP |
GO:0006942-Regulation of striated muscle
contraction | 5 | 3.56E-5 |
| KEGG pathway | Hsa04020: Calcium
signaling pathway | 8 | 1.40E-3 |
| KEGG pathway | Hsa04960:
Aldosterone-regulated sodium reabsorption | 4 | 7.30E-3 |
| KEGG pathway | Hsa04270: Vascular
smooth muscle contraction | 5 | 2.36E-2 |
| KEGG pathway | Hsa04920:
Adipocytokine signaling pathway | 4 | 2.75E-2 |
| KEGG pathway | Hsa04260: Cardiac
muscle contraction | 4 | 4.06E-2 |
Construction of the PPI network
The PPI network was constructed using all DEGs and
included 292 nodes (individual proteins) and 731 edges (interaction
events), as shown in Fig. 2. In
the PPI network, MYC interacted with MAP3K14 and GTPBP4. The
interacting proteins of BYSL included GTPBP4, NOP14, NOP2, NOB1,
AATF and TRMT6. Following analysis of the network topology, eight
DEGs (degree >20) were identified (Table III), including MYC (degree=29),
NOP2 (degree=29), GTPBP4 (degree=27), BYSL (degree=25) and AATF
(degree=22). The degree represents the number of interactions with
other proteins.
| Table III.Eight differentially expressed genes
(degree >20) in the network topology. |
Table III.
Eight differentially expressed genes
(degree >20) in the network topology.
| Gene | Degree | Betweenness | Closeness |
|---|
| MYC | 29.0 | 14,147.05 | 0.054 |
| NOP2 | 29.0 |
4,228.61 | 0.053 |
| GTPBP4 | 27.0 |
5,434.96 | 0.053 |
| ACACB | 25.0 | 12,036.69 | 0.053 |
| BYSL | 25.0 |
1,432.08 | 0.052 |
| MYH6 | 23.0 |
5,115.25 | 0.053 |
| AATF | 22.0 |
4,423.36 | 0.053 |
| NAT10 | 22.0 |
1,173.44 | 0.052 |
Function of the PPI network
module
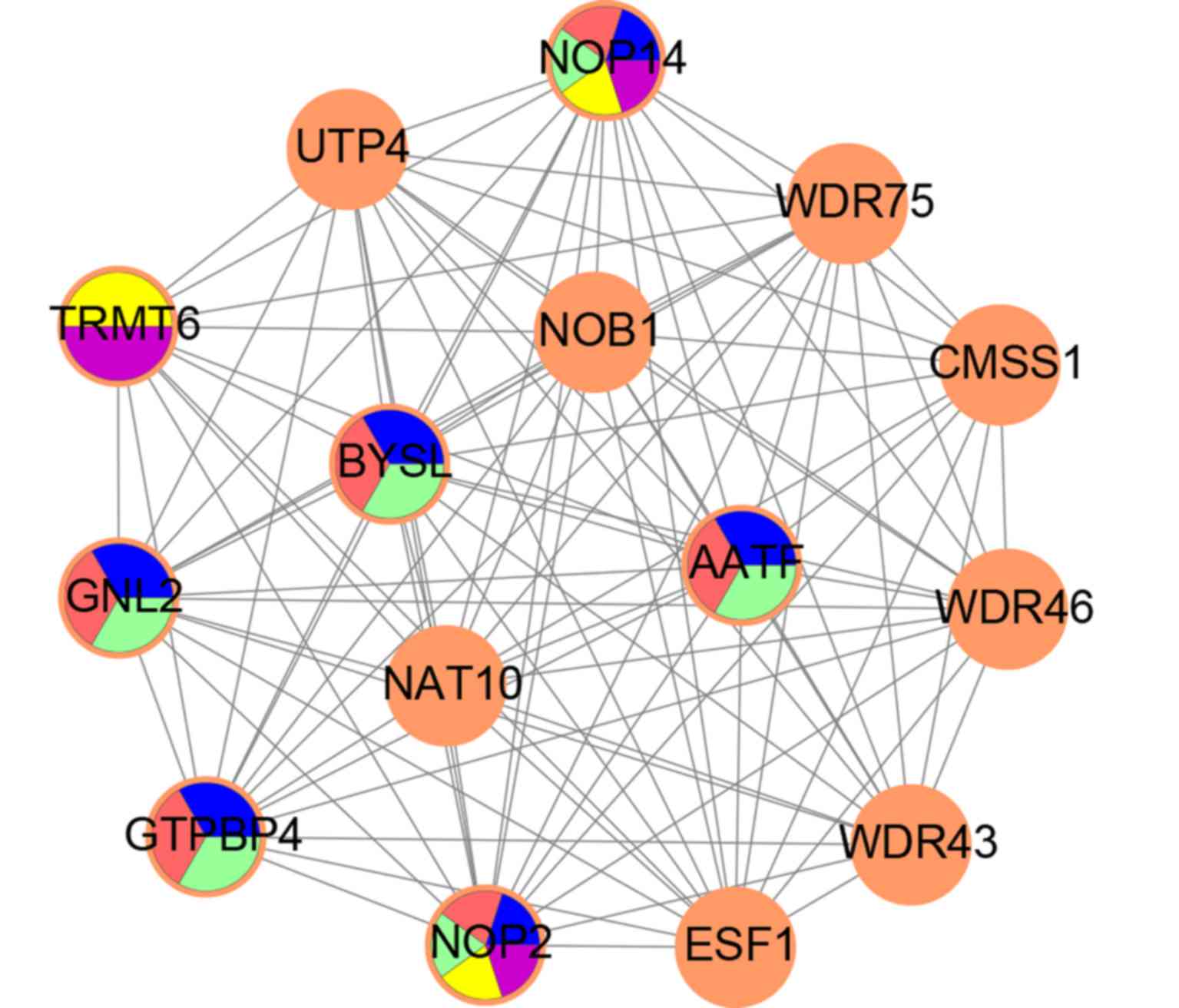
The MCODE module with the highest score (14.286) in
the PPI network was identified; it included 15 upregulated DEGs and
100 edges. The function of this module was enriched in ribosome
biogenesis following GO analysis (Fig.
3). The top five GO terms in the module were analyzed using GO
analysis, and the results showed that NOP14, NOP2, AATF, GNL2,
GTPBP4, BYSL and TRMT6 were key genes in the module (Fig. 4).
Construction of the integrated
DEG-miRNA-TF regulatory network
The integrated regulatory network was constructed
with the target DEGs, miRNAs and TFs (Fig. 5). In the network, hsa-miR-150 was
identified, which was the only miRNA associated with sepsis. Its
target DEGs, including MYLK3, are shown in Table IV. A total of 21 TFs were
identified in the network, comprising 14 upregulated DEGs,
including MYC, WDR46, IPO4 and ZNF593, and seven
downregulated DEGs (Table V).
| Table IV.miRNA-target DEGs in the integrated
DEG-miRNA-TF regulatory network. |
Table IV.
miRNA-target DEGs in the integrated
DEG-miRNA-TF regulatory network.
| miRNA | Target genes |
|---|
| Hsa-miR-150-3p | SRXN1,
DHX33 |
| Hsa-miR-150-5p | AGMAT, PPP2R3A,
MYLK3, PNPLA3, XPOT, PAIP2B |
| Table V.TFs in the integrated DEG-miRNA-TF
regulatory network. |
Table V.
TFs in the integrated DEG-miRNA-TF
regulatory network.
| DEGs | TFs |
|---|
| Upregulated | DDX21, FKBP5,
HMGB3, IPO4, KPNA2, MYC, NAT10, PRMT3, SSRP1, TTC13, WDR43, WDR46,
WDR75, ZNF593 |
| Downregulated | COL15A1, CSRP3,
E2F8, FBXO40, LDB3, NR3C2, NRAP |
Discussion
In the present study, there were 259 upregulated and
204 downregulated DEGs in the samples from patients with sepsis,
compared with those in samples from healthy controls. The
upregulated DEGs were enriched in the apoptosis pathway and in the
metabolism of glutathione. The downregulated DEGs were enriched in
pathways, including the calcium signaling pathway and adipocytokine
signaling pathway. A total of 292 nodes and 731 edges were
identified in the PPI network, including eight DEGs with higher
degrees (degree >20). Subsequently, key genes in the module with
the maximum score (14.286; 15 upregulated DEGs and 100 edges) in
the PPI network were identified. Finally, target DEGs, miRNAs and
TFs were identified in samples from patients with sepsis.
In the PPI network, MYC, NOP2, GTPBP4, BYSL and AATF
had a higher degree (>20). The MYC protein is encoded by the
MYC gene, which is associated with cell cycle,
transformation and apoptosis (27). There is no direct evidence to
confirm the role of MYC in sepsis. However, proinflammatory
cytokines, including TNF-α, are closely associated with sepsis
characterized by SIRS (28), which
may have important implications on the treatment of sepsis
(29). For example, TNF-α protects
against microvascular endothelial dysfunction in patients with
sepsis by activating NF-B and p38 in endothelial cells (30). In addition, MYC promotes the
expression of TNF-α in chronic liver diseases (31). Therefore, MYC may be crucial in the
progress of sepsis by promoting the expression of TNF-α. Qiao et
al found that NOP2 and GTPBP4 may be
differentially expressed in neonatal sepsis (32), which is consistent with the results
of the present study. Therefore, NOP2 and GTPBP4 may be associated
with the progress of sepsis. The AATF protein, also known as Che-1,
is encoded by the AATF gene, which is involved in cell apoptosis
(33). The overexpression of AATF
interferes with MAP3K12/DLK (a protein kinase)-induced apoptosis
(34,35), which forms heterodimers with a
leucine zipper containing TFs, including MYC, and has a regulatory
role (36). There is no evidence
to confirm the function of AATF in sepsis. However,
AATF represents an immune- or inflammation-associated gene
and encodes miRNA-2909, which is responsible for the regulation of
genes involved in inflammation (37,38).
Therefore, AATF may be associated with inflammation in sepsis by
regulating the expression of miRNA-2909.
BYSL, an adhesion molecule in mammalian
preimplantation embryos (39), is
crucial in pre-ribosomal RNA (rRNA) processing, and its depletion
leads to the accumulation of an unusual 18S rRNA precursor
(40). There is no evidence to
confirming its association with sepsis. In the present study, BYSL
was found to closely interact with GTPBP4, NOP14, NOP2, NOB1 and
AATF in the PPI network. In addition, the NOP14 gene encodes
a NOP14 protein, which is involved in pre-18S rRNA processing and
small ribosomal subunit assembly (41). NOB1 is involved in pre-rRNA
processing and produces a mature 18S rRNA in a late cytoplasmic
processing step (42). Studies
have reported that 16S rRNA gene amplification by real-time
polymerase chain reaction and sequencing rapidly and accurately
diagnose neonatal sepsis (43–45).
Therefore, BYSL, NOB1 and NOP14 are involved in
pre-18S rRNA processing in sepsis.
In the present study, hsa-miR-150, including its
target genes (MYLK3) was identified in the integrated
DEG-miRNA-TF regulatory network. Zhao et al also found that
the serum levels of miR-150 decreased in a rat sepsis model,
providing a valuable tool in evaluating the prognosis of sepsis
(11). Similarly, the expression
of miR-150 was reported to be decreased in a patient with sepsis
(46). A previous study found that
MLCK, including MYLK1, MYLK2 and MYLK3, may
not upregulate endothelial permeability induced by LPS exposure,
whereas hyper-permeability was observed in sepsis and other
inflammatory situations (47).
TNF-α, LPS and 18% cyclic stretch each increased the activity of a
MYLK gene 3′untranslated region luciferase reporter, and
induction was reduced by mimics of each miRNA (48). As reported in the above study, a
negative correlation was found between the expression of miR-150
and white blood cell counts, and the levels of IL-6, C-reactive
protein and TNF-α (11). In the
present study, MYLK3 was identified as a downregulated DEG
in sepsis and one of the target genes of miR-150. These findings
suggested that the expression of miR-150 is increased by
upregulating MYLK3, which provides a direction for sepsis
therapy by developing a novel drug targeting MYLK3 in
miR-150.
There were a number of limitations in the present
study, with the exception of the above finding. All predicted
results require confirmation by laboratory data, however, there are
currently insufficient samples from patients with sepsis. In future
investigations, the expression of the DEGs discussed above requires
validation using large-scale samples. In addition, future
investigations aim to confirm the interactions of DEGs, regulatory
associations between TFs and DEGs, and possible pathways underlying
these gene alterations.
In conclusion, a total of 259 upregulated DEGs,
including MYC, AATF and BYSL, and 204 downregulated
DEGs, including MYLK3, were identified from patients with
sepsis. NOP14, NOP2, AATF, GTPBP4, BYSL, MYC and
MYLK3 were key genes in sepsis, which may be involved in
pre-18S rRNA processing in sepsis or may be associated with
inflammation in sepsis by regulating the expression of miRNA-2909.
In addition, miR-150 and its target genes, including MYLK3,
were found to be key in sepsis. The findings may provide further
understanding of the pathogenesis of sepsis and assist in
developing a novel target drug for the treatment of sepsis.
Glossary
Abbreviations
Abbreviations:
|
miRNA
|
microRNA
|
|
DEGs
|
differentially expressed genes
|
|
DAVID
|
database for annotation, visualization
and integration discovery
|
|
GEO
|
gene expression omnibus
|
|
PPI
|
protein-protein interaction
|
|
LIMMA
|
linear models for microarray
|
|
TFs
|
transcription factors
|
|
SIRS
|
systemic inflammatory response
syndrome
|
|
IL-6
|
interleukin-6
|
|
TNF-α
|
tumor necrosis factor-α
|
|
LPS
|
lipopolysaccharides
|
|
GO
|
gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
FC
|
fold change
|
References
|
1
|
Tsai D, Stewart P, Goud R, Gourley S,
Hewagama S, Krishnaswamy S, Wallis SC, Lipman J and Roberts JA:
Total and unbound ceftriaxone pharmacokinetics in critically ill
Australian Indigenous patients with severe sepsis. Int J Antimicrob
Agents. 48:748–752. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleischmann C, Scherag A, Adhikari NK,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K:
International Forum of Acute Care Trialists: Assessment of Global
incidence and mortality of Hospital-treated Sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng B, Xie G, Yao S, Wu X, Guo Q, Gu M,
Fang Q, Xu Q, Wang D, Jin Y, et al: Epidemiology of severe sepsis
in critically ill surgical patients in ten university hospitals in
China. Crit Care Med. 35:2538–2546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wallisch JS, Pang D, Carcillo JA and Aneja
RK: Implementation of guidelines to treat pediatric sepsis:
Cookbook medicine or the force awakens! Pediatr Crit Care Med.
17:884–885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baghel K, Srivastava RN, Chandra A, Goel
SK, Agrawal J, Kazmi HR and Raj S: TNF-α, IL-6, and IL-8 cytokines
and their association with TNF-α-308 G/A polymorphism and
postoperative sepsis. J Gastrointest Surg. 18:1486–1494. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thimmulappa RK, Lee H, Rangasamy T, Reddy
SP, Yamamoto M, Kensler TW and Biswal S: Nrf2 is a critical
regulator of the innate immune response and survival during
experimental sepsis. J Clin Invest. 116:984–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Coquerel D, Neviere R, Delile E, Mulder P,
Marechal X, Montaigne D, Renet S, Remy-Jouet I, Gomez E, Henry JP,
et al: Gene deletion of protein tyrosine phosphatase 1B protects
against sepsis-induced cardiovascular dysfunction and mortality.
Arterioscler Thromb Vasc Biol. 34:1032–1044. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kloosterman WP, Wienholds E, de Bruijn E,
Kauppinen S and Plasterk RH: In situ detection of miRNAs in animal
embryos using LNA-modified oligonucleotide probes. Nat Methods.
3:27–29. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cifuentes D, Xue H, Taylor DW, Patnode H,
Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al: A
Novel miRNA processing pathway independent of dicer requires
Argonaute2 catalytic activity. Science. 328:1694–1698. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goodwin AJ, Guo C, Cook JA, Wolf B,
Halushka PV and Fan H: Plasma levels of microRNA are altered with
the development of shock in human sepsis: An observational study.
Crit Care. 19:4402015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao X, Wang Y and Chen Z: The predict
value of serum miRNA150 on sepsis prognosis. Int J Clin Exp Med.
9:18367–18372. 2016.
|
|
12
|
Fredriksson K, Tjäder I, Keller P,
Petrovic N, Ahlman B, Schéele C, Wernerman J, Timmons JA and
Rooyackers O: Dysregulation of mitochondrial dynamics and the
muscle transcriptome in ICU patients suffering from sepsis induced
multiple organ failure. PLoS One. 3:e36862008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fredriksson K, Tjäder I, Keller P,
Petrovic N, Ahlman B, Schéele C, Wernerman J, Timmons JA and
Rooyackers O: Dysregulation of mitochondrial dynamics and the
muscle transcriptome in ICU patients suffering from sepsis induced
multiple organ failure. PLoS One. 3:e36862008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ogata H, Goto S, Sato K, Fujibuchi W, Bono
H and Kanehisa M: KEGG: Kyoto encyclopedia of genes and genomes.
Nucleic Acids Res. 27:29–34. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
von Mering C, Huynen M, Jaeggi D, Schmidt
S, Bork P and Snel B: STRING: A database of predicted functional
associations between proteins. Nucleic Acids Res. 31:258–261. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of protein interaction networks. Biosystems. 127:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Garcia O, Saveanu C, Cline M,
Fromont-Racine M, Jacquier A, Schwikowski B and Aittokallio T:
GOlorize: A cytoscape plug-in for network visualization with Gene
Ontology-based layout and coloring. Bioinformatics. 23:394–396.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dweep H and Gretz N: miRWalk2.0: A
comprehensive atlas of microRNA-target interactions. Nat Methods.
12:6972015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng G, Tu K, Yang Q, Xiong Y, Wei C, Xie
L, Zhu Y and Li Y: ITFP: An integrated platform of mammalian
transcription factors. Bioinformatics. 24:2416–2417. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matys V, Kel-Margoulis OV, Fricke E,
Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M,
Hornischer K, et al: TRANSFAC and its module TRANSCompel:
Transcriptional gene regulation in eukaryotes. Nucleic Acids Res.
34:(Database Issue). D108–D110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koh CM, Sabò A and Guccione E: Targeting
MYC in cancer therapy: RNA processing offers new opportunities.
Bioessays. 38:266–275. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roderburg C, Benz F, Schüller F, Pombeiro
I, Hippe HJ, Frey N, Trautwein C, Luedde T, Koch A, Tacke F and
Luedde M: Serum levels of TNF receptor ligands are dysregulated in
sepsis and predict mortality in critically Ill patients. PLoS One.
11:e01537652016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hotchkiss RS, Monneret G and Payen D:
Immunosuppression in sepsis: A novel understanding of the disorder
and a new therapeutic approach. Lancet Infect Dis. 13:260–268.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang Y, Li X, Zhang X, Li Z, Wang L, Sun
Y, Liu Z and Ma X: Elevated levels of plasma TNF-α are associated
with microvascular endothelial dysfunction in patients with sepsis
through activating the NF-κB and p38 mitogen-activated protein
kinase in endothelial cells. Shock. 41:275–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu T, Zhou Y, Ko KS and Yang H:
Interactions between Myc and mediators of inflammation in chronic
liver diseases. Mediators Inflamm. 2015:2768502015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qiao X, Zhu S, Zhang S and Dong H:
Disrupted pathways associated with neonatal sepsis: Combination of
protein-protein interactions and pathway data. BioChip J. 11:1–7.
2017. View Article : Google Scholar
|
|
33
|
Liu M, Wang D and Li N: Che-1 gene
silencing induces osteosarcoma cell apoptosis by inhibiting mutant
p53 expression. Biochem Biophys Res Commun. 473:168–173. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Smigiel R, Marcelis C, Patkowski D, de
Leeuw N, Bednarczyk D, Barg E, Mascianica K, Maria Sasiadek M and
Brunner H: Oesophageal atresia with tracheoesophageal fistula and
anal atresia in a patient with a de novo microduplication in 17q12.
Eur J Med Genet. 57:40–43. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
De Nicola F, Bruno T, Iezzi S, Di Padova
M, Floridi A, Passananti C, Del Sal G and Fanciulli M: The prolyl
isomerase Pin1 affects Che-1 stability in response to apoptotic DNA
damage. J Biol Chem. 282:19685–19691. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baker ST, Opperman KJ, Tulgren ED, Turgeon
SM, Bienvenut W and Grill B: RPM-1 uses both ubiquitin ligase and
phosphatase-based mechanisms to regulate DLK-1 during neuronal
development. PLoSGenet. 10:e10042972014.
|
|
37
|
Sharma S, Singh D and Kaul D: AATF RNome
has the potential to define post mortem interval. Forensic Sci Int.
247:e21–e24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SJ, Lee EJ, Kim SK, Jeong P, Cho YH,
Yun SJ, Kim S, Kim GY, Choi YH, Cha EJ, et al: Identification of
pro-Inflammatory cytokines associated with muscle invasive bladder
cancer; the roles of IL-5, IL-20, and IL-28A. PLoS One.
7:e402672012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hashimoto M, Sato T, Muroyama Y, Fujimura
L, Hatano M and Saito T: Nepro is localized in the nucleolus and
essential for preimplantation development in mice. Dev Growth
Differ. 57:529–538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Carron C, O'Donohue MF, Choesmel V,
Faubladier M and Gleizes PE: Analysis of two human pre-ribosomal
factors, bystin and hTsr1, highlights differences in evolution of
ribosome biogenesis between yeast and mammals. Nucleic Acids Res.
39:280–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu PC and Thiele DJ: Novel
stress-responsive genes EMG1 and NOP14 encode conserved,
interacting proteins required for 40S ribosome biogenesis. Mol Biol
Cell. 12:3644–3657. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang J, McCann KL, Chen Q, Gonzalez LE,
Baserga SJ and Hall TM: Nop9 is a PUF-like protein that prevents
premature cleavage to correctly process pre-18S rRNA. Nat Commun.
7:130852016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
El Gawhary S, El-Anany M, Hassan R, Ali D
and El Gameel el Q: The role of 16S rRNA gene sequencing in
confirmation of suspected neonatal sepsis. J Trop Pediatr.
62:75–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mithal LB, Malczynski M, Green SJ, Qi C,
Yogev R and Mestan K: Deep sequencing of 16S rRNA gene amplicons to
screen umbilical cord blood of preterm infants. Open Forum
Infectious Dis. 3:22342016.
|
|
45
|
Midan DA, Abo El Fotoh WMM and El
Shalakany AH: The potential role of incorporating real-time PCR and
DNA sequencing for amplification and detection of 16S rRNA gene
signatures in neonatal sepsis. J Matern Fetal Neonatal Med.
30:1476–1483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ticlea M, Bratu LM, Bodog F, Bedreag OH,
Rogobete AF and Crainiceanu ZP: The use of exosomes as biomarkers
for evaluating and monitoring critically Ill polytrauma patients
with sepsis. Biochem Genet. 55:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu H, Yu X, Yu S and Kou J: Molecular
mechanisms in lipopolysaccharide-induced pulmonary endothelial
barrier dysfunction. Int Immunopharmacol. 29:937–946. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Adyshev DM, Moldobaeva N, Mapes B,
Elangovan V and Garcia JG: MicroRNA regulation of nonmuscle myosin
light chain kinase expression in human lung endothelium. Am J
Respir Cell Mol Biol. 49:58–66. 2013. View Article : Google Scholar : PubMed/NCBI
|