Introduction
Angiomyolipoma (AML) is a benign mesenchymal
neoplasm composed of smooth muscle cells, adipose tissue and
thick-walled dystrophic blood vessels arising from perivascular
epithelioid cells. Epithelioid AML (EAML) is a variant of AML,
which is characterized by more aggressive growth and a less
favorable clinical outcome. EAML is composed of cells arranged in
nests, sheets or broad alveoli surrounded by vascular septae. The
cells within the tumor may be spindled or epithelioid, with clear
or eosinophilic cytoplasm. There are also tumors that present with
numerous pleomorphic multinucleated cells. Cases of local
recurrence and metastasis, mainly in the liver, lymph nodes, lungs,
other retroperitoneal organs and bones, have been previously
described. The histopathological diagnosis of this tumor may be
difficult, as EAML often mimics other neoplasms (1–5). This is
the case report of a 39-year-old male patient with EAML, which was
initially diagnosed as adrenal cortical carcinoma (ACC), due to the
lack of proper cooperation between clinicians and pathologists.
Case report
Patient
In December 2013, a 39-year-old male patient
diagnosed with cancer of the right adrenal gland was admitted to
the Department of Endocrinology, Metabolism and Internal Medicine,
Poznań University of Medical Sciences (Poznań, Poland) for further
treatment.
Medical interview
The patient presented 3 years prior with a tumor in
the right perirenal area, which was resected and diagnosed as a
neoplasm of uncertain origin, possibly AML or metastatic melanoma.
Following surgery, the patient underwent 6 months of oncological
follow-up. In September, 2013 the patient was admitted to the
hospital with low-grade fever and abdominal pain. A computed
tomography (CT) scan of the abdomen revealed a large
retroperitoneal tumor in close proximity to the right kidney. The
patient underwent surgery in October, 2013 and the tumor along with
the right kidney were resected.
Macroscopic findings
A tissue specimen described as an adrenal gland
tumor measuring 16.0×9.0×10.0 cm was sent to the Department of
Pathology (Poznań University of Medical Sciences). On sectioning,
the tumor was polycystic, tan-brown, including a prominent necrotic
and hemorrhagic area measuring 10.0×8.0×6.0 cm. A fragment of
orange-colored tissue was identified, measuring 1.1×1.5×0.7 cm,
which was considered to be residual adrenal gland tissue. The tumor
infiltrated through the capsule of the kidney, whereas the
remainder of the kidney, renal pelvis, ureter and blood vessels
presented no significant macroscopic findings.
Microscopic findings
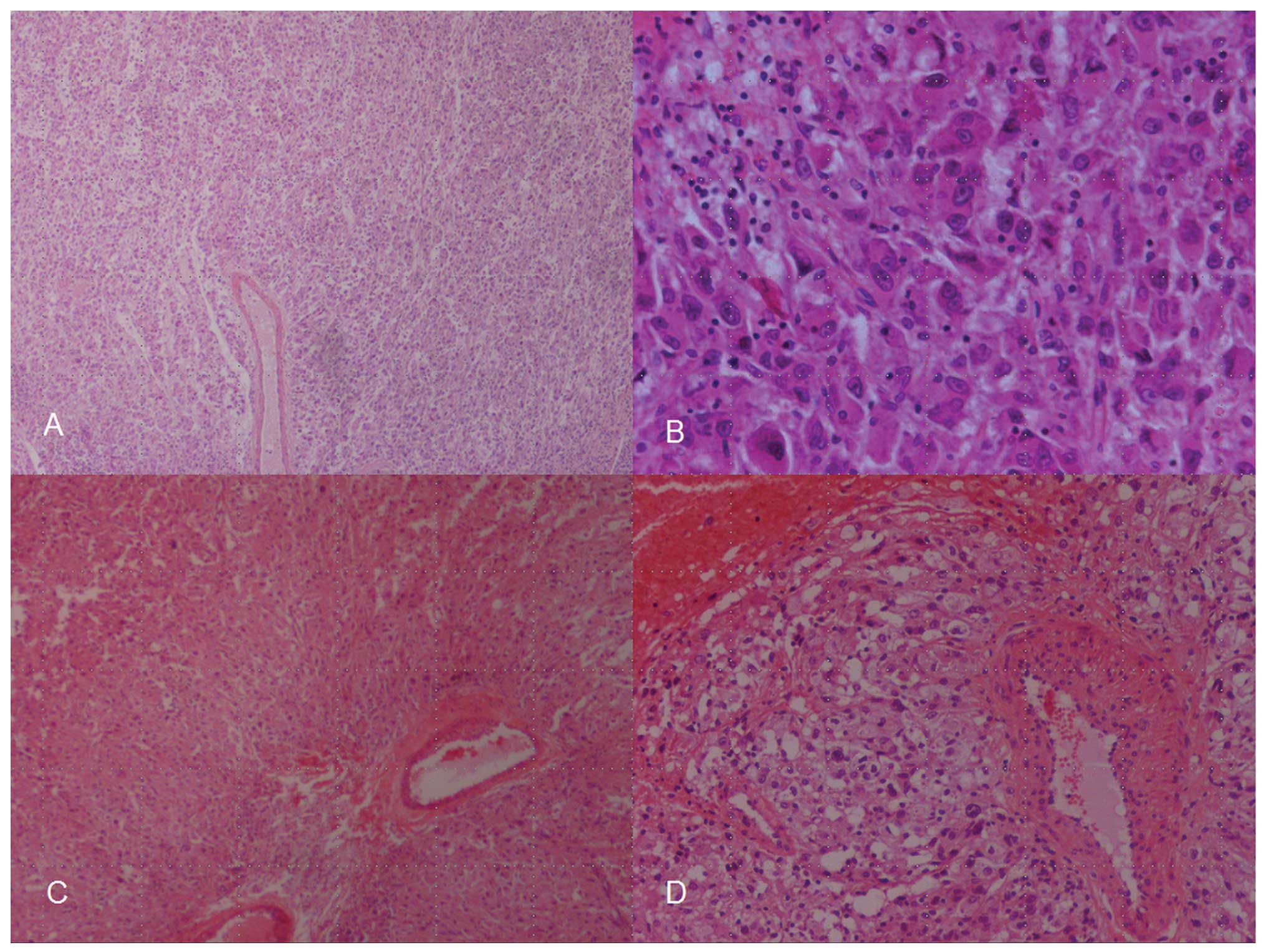
The histopathology report described a tumor composed
mostly of epithelioid cells with high nuclear pleomorphism, which
formed solid areas and smaller nests surrounded by numerous blood
vessels. The tumor infiltrated the capsule and cortex of the kidney
and the surgical margins were positive for tumor cells. Necrotic
and hemorrhagic areas were identified within the tumor, whereas the
mitotic index was 5–7 mitoses per 10 high-power fields (Fig. 1). The tumor exhibited a strong
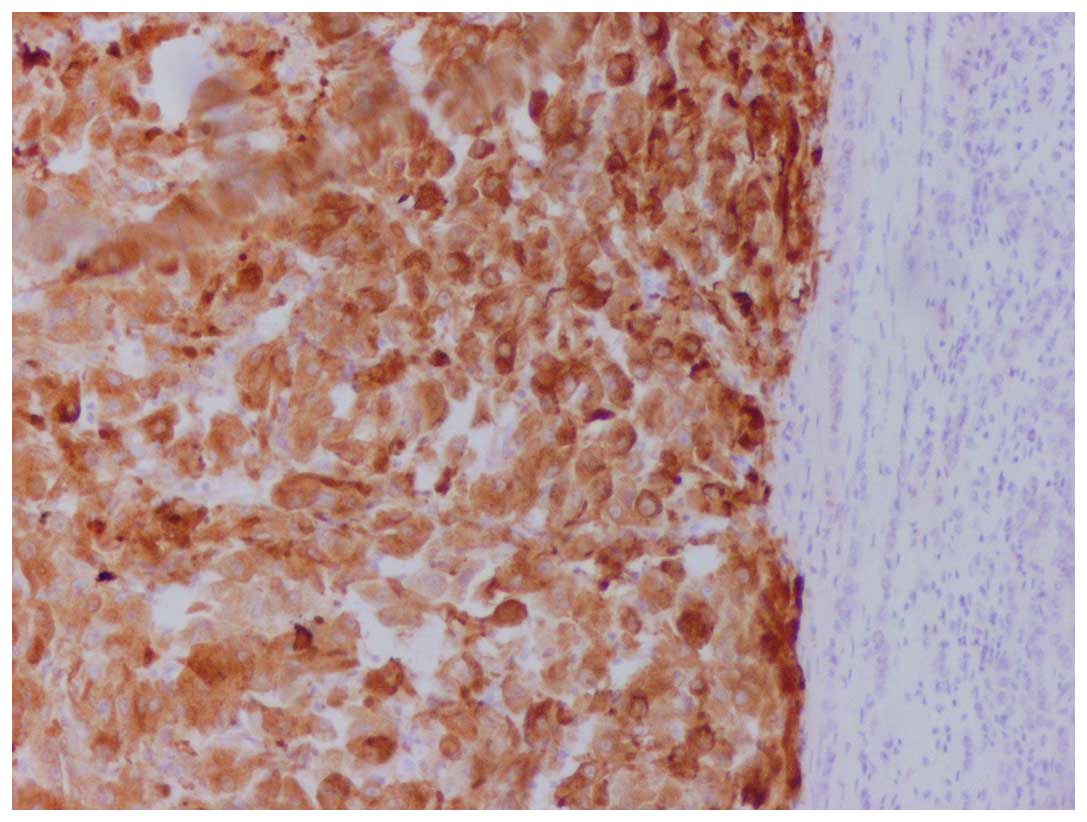
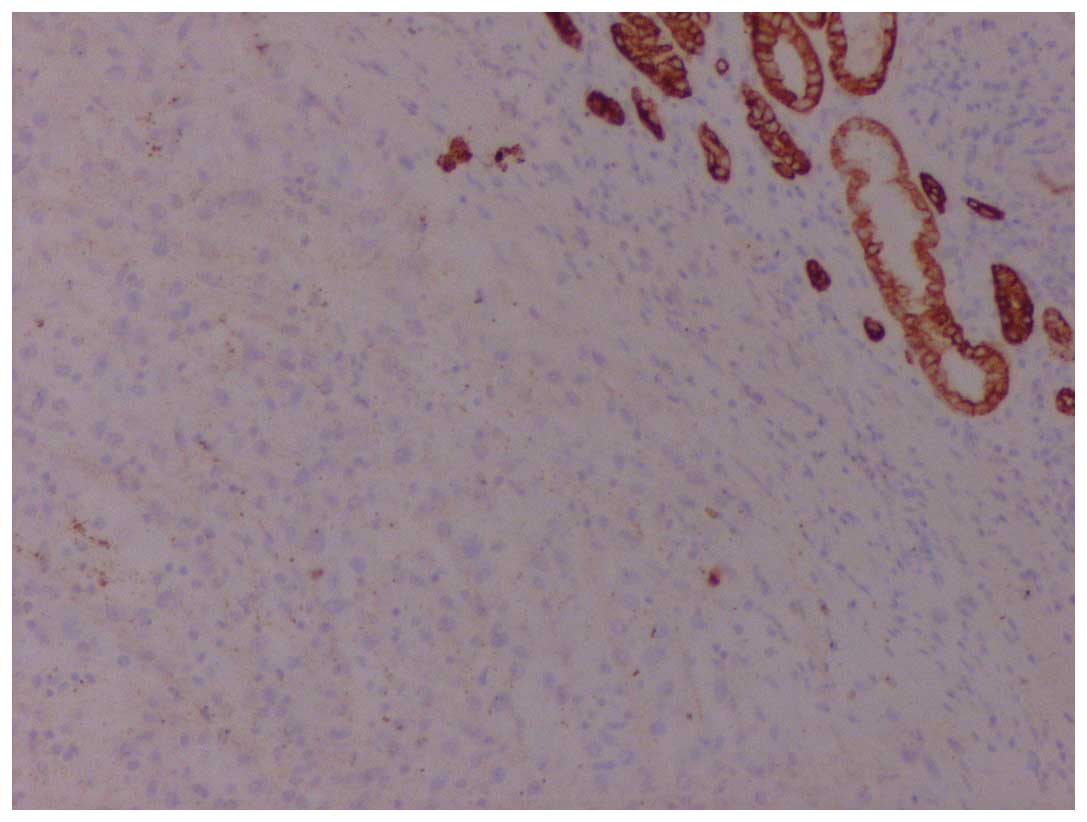
immunoreactivity for vimentin, human melanoma black-45 (Fig. 2) and Melan-A (Fig. 3), whereas it was negative for keratins
(Fig. 4), S100, synaptophysin and
chromogranin A. The histopathology report concluded that, despite
the mildly atypical immunohistochemical examination results, the
microscopic characteristics strongly suggested the diagnosis of
ACC. This diagnosis was also supported by the fact that the tumor
was sent to the Department of Pathology as an adrenal tumor and the
gross examination also strongly suggested that it originated from
the adrenal gland.
The patient was diagnosed with adrenal gland
carcinoma and was scheduled to receive mitotane treatment. Prior to
the onset of treatment, the detailed medical history of the patient
was recorded and all the necessary tests and examinations were
performed. The patient had no known family history of endocrine
malignancies.
Physical examination
Upon admission, the general condition of the patient
was good, with the exception of complaints regarding minor pain in
the muscles of the upper extremities. On physical examination there
were no significant findings, such as signs of hypercortisolemia.
The laboratory tests revealed dyslipidemia, marginally elevated
creatinine levels and iron deficiency. The radiological imaging
findings were unremarkable, revealing intact bilateral adrenal
glands and absence of the right kidney. On bone scintigraphy
analysis, there was increased tracer concentration in the left
humeral joint (there was a query regarding previous arm luxation)
and focal changes in the ribs suggestive of either osteoporotic
fractures or metastases.
Due to the unexpectedly good condition of the
patient, reassessment of the initial adrenal cancer diagnosis was
considered, starting with the re-evaluation of the pathological
findings. The pathologists were informed that the patient underwent
surgery for a kidney tumor in 2010 in another hospital, and that
the specimen sent to the Department of Pathology was a kidney tumor
rather than an adrenal gland tumor. The pathologists also retrieved
the previous histopathology report. Taking into consideration all
the available information, the pathologists decided to perform
additional immunohistochemical analyses, ultimately changing the
previous diagnosis of adrenal gland carcinoma to EAML. Considering
that this was a recurrent tumor with a diameter of >10 cm,
composed mostly of atypical epithelioid cells with increased
mitotic activity and infiltration of the perinephric fat, the
pathologists were unable to predict the benign or malignant
behavior of the tumor, as such lesions may recur and even
metastasize. The patient was discharged in a good general
condition. Further oncological follow-up was recommended.
Written informed consent for the publication of this
case report was obtained from the patient.
Discussion
The epithelioid variant of AML was first described
in 1995. According to the available data, only ~160 cases of EAML
had been described up to 2014. There are several characteristics
distinguishing EAML from classic AML. The majority of EAMLs are
larger compared with AMLs (the average size of the tumor at
diagnosis has been reported to be 8.6 cm) (3), are mainly composed of epithelioid cells,
and often lack the typical fat tissue component characteristic for
this type of neoplasm. The malignant behavior, recurrence and
metastasis rates of EAMLs cannot be easily determined. The average
age at diagnosis of AML is 40 years (6,7).
EAML may be difficult to diagnose, as it may mimic
other neoplasms on radiological imaging and histopathological
examination. EAML is known to display a variable
immunohistochemical phenotype and, therefore, may be misdiagnosed
as renal cell carcinoma, ACC, hepatocellular carcinoma, epithelioid
variant of malignant melanoma or hepatoblastoma. A comparison of
the immunohistochemical profile of EAML, the present case and ACC
is presented in Table I (3,8,9).
 | Table I.Immunohistochemical comparison of
EAML, the present case and ACC. |
Table I.
Immunohistochemical comparison of
EAML, the present case and ACC.
| Immunohistochemical
markers | EAML | Present case | ACC |
|---|
| S-100 | −/+ | – | +/− |
| Melan-A | + | + | + |
| HMB-45 | + | + | – |
| CD117 | + | – | −/+ |
| CD63 | + | Not performed | NR |
| Keratins | −/+ | – | −/+ |
| SMA | +/- | +a | – |
| Desmin | −/+ | –a | – |
| Vimentin |
| + | + |
| Synaptophysin |
| – | + |
| Calretinin |
|
| + |
| D2-40 |
|
| + |
| Inhibin |
|
| + |
| SF1 |
|
| + |
| Pankeratin |
|
| +/- |
The coexistence of EAML with tuberous sclerosis is
the most essential characteristic of its clinical presentation.
Tuberous sclerosis is a genetic disorder characterized by mental
impairment, autism, seizures and the presence of various tumors. In
certain cases, the disease is otherwise asymptomatic, apart from
the mass effect of the tumor as it grows to a larger size (10).
According to Nese et al (7), the coexistence of EAML with tuberous
sclerosis or AML, the presence of tumor necrosis, infiltration of
the tumor beyond the renal parenchyma, infiltration of the renal
vein and ‘carcinoma-like’ growth pattern are characteristics
associated with an unfavorable prognosis. The main treatment is the
resection of the tumor. Alternative treatment strategies include
chemoradiation or arterial embolization of the tumor. There have
also been attempts to use mammalian target of rapamycin (mTOR)
inhibitors, including temsirolimus and everolimus (11).
ACC is a rare epithelial tumor, which is derived
from the cortex of the adrenal gland, with a frequency of 1–12
cases per million (12–14). ACC most commonly occurs in patients
aged 40–60 years and in children aged <5 years (12,14) and
its mean diameter ranges between 5 and 20 cm (14). ACC is usually a sporadic tumor;
however, it may occasionally be associated with the Li-Fraumeni
syndrome, multiple neuroendocrine neoplasia type 1, or the Carney
complex (14). The clinical
manifestations of this neoplasm depend on its hormonal activity,
which may be present in 60–80% of adult patients displaying
symptoms of Cushing's syndrome or androgenization (14). In rare cases of ACC without hormonal
activity, the symptoms are associated with the presence of the
tumor mass or the presence of distant metastases and disseminated
neoplastic disease (14). There are
four clinical stages of ACC (15).
The histopathological diagnosis of ACC may be
difficult. There is currently no consensus regarding
decision-making as to whether ACCs are benign or malignant. The
diagnosis is most commonly based on the Weiss criteria with
Aubert's modifications (Table II)
(16).
| Table II.Weiss criteria with Aubert's
modifications. The threshold for identifying malignant behaviour is
≥3. |
Table II.
Weiss criteria with Aubert's
modifications. The threshold for identifying malignant behaviour is
≥3.
| No. | Criteria |
|---|
| 1 | Nuclear grade by
Fuhrman (III/IV) |
| 2 | Mitotic index
(>5/50 high-power fields) |
| 3 | Atypical mitoses |
| 4 | Clear cells
(<25%) |
| 5 | Diffuse architecture
(>33%) |
| 6 | Necrosis |
| 7 | Venus invasion |
| 8 | Sinusoidal
invasion |
| 9 | Capsular
invasion |
The treatment of ACC includes surgery and mitotane
therapy, which is commonly associated with side effects, requiring
a reduction of the dose. In particular cases, it may be necessary
to add chemotherapy with etoposide, doxorubicin and cisplatin or
streptokinase. The prognosis for patients with ACC is unfavorable
(17).
The diagnosis of EAML and ACC may be challenging.
Cases of stage-1 and −2 ACC without hormonal activity may be
clinically identical to EAML. In the present case, the age of the
patient was not a valuable indicator, as it was typical for both
types of neoplasm.
It was significant for the endocrinologist that the
general health condition of the patient was surprisingly good,
despite the fact that the diagnosis was ACC relapse. There were
also doubts regarding the results of the histopathological
examination after the first operation. Additionally, the presence
of both adrenal glands on CT imaging caused the clinician to
consider verifying the second histopathological diagnosis.
The present case stresses the significance of the
close cooperation between clinicians and pathologists. It is
necessary to provide pathologists with all the available medical
history and physical examination results. In the present case, the
pathologist was wrongly informed of the tumor site (the tumor was
previously described and sent to the Department of Pathology as an
adrenal neoplasm). There was also no information regarding the
previous histology results, which considered the diagnosis of EAML
or metastatic melanoma. This information would not only help the
pathologist reach an accurate diagnosis, but would also save time
and money spent on immunohistochemical assays. The thorough review
of the medical history of the patient by the clinician helped avoid
further unnecessary toxic and costly treatment.
Abbreviations:
|
AML
|
angiomyolipoma
|
|
EAML
|
epithelioid angiomyolipoma
|
|
ACC
|
adrenal cortical carcinoma
|
References
|
1
|
Martignoni G and Amin MB: Angiomyolipoma.
In: WHO Classification of Tumours. Pathology and GeneticsTumours of
the Urinary System and Male Genital Organs. Eble JN, Sauter G,
Epstein JI and Sesterhenn IA: IARC Press; Lyon, France: pp. 63–67.
2004
|
|
2
|
Amin MB: Epithelioid angiomyolipoma. In:
WHO Classification of Tumours. Pathology and GeneticsTumours of the
Urinary System and Male Genital Organs. Eble JN, Sauter G, Epstein
JI and Sesterhenn IA: IARC Press; Lyon, France: pp. 68–69. 2004
|
|
3
|
Mete O and van der Kwast TH: Epithelioid
angiomyolipoma: A morphologically distinct variant that mimics a
variety of intra-abdominal neoplasms. Arch Pathol Lab Med.
135:665–670. 2011.PubMed/NCBI
|
|
4
|
Eble JN, Amin MB and Young RH: Epithelioid
angiomyolipoma of the kidney: A report of five cases with a
prominent and diagnostically confusing epithelioid smooth muscle
component. Am J Surg Pathol. 21:1123–1130. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hornik JL and Fletcher CD: PEComa: What do
we know so far? Histopathology. 48:75–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nelson CP and Sanda MG: Contemporary
diagnosis and management of renal angiomyolipoma. J Urol.
168:1315–1325. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nese N, Martignoni G, Fletcher CD, et al:
Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of
the kidney: A clinicopathologic study of 41 cases: Detailed
assessment of morphology and risk stratification. Am J Surg Pathol.
35:161–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sangoi AR, Fujiwara M, West RB, Montgomery
KD, Bonventre JV, Higgins JP, Rouse RV, Gokden N and McKenney JK:
Immunohistochemical distinction of primary adrenal cortical lesions
from metastatic clear cell renal cell carcinoma: A study of 248
cases. Am J Surg Pathol. 35:678–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fulawka L, Patrzalek D and Halon A:
Adrenal cortical carcinoma with extension into the inferior vena
cava - case report and literature review. Diagn Pathol. 9:512014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lane BR, Aydin H, Danforth TL, Zhou M,
Remer EM, Novick AC and Campbell SC: Clinical correlates of renal
angiomyolipoma subtypes in 209 patients: classic, fat poor,
tuberous sclerosis associated and ephitelioid. J Urol. 180:836–843.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wyluda E, Baqero G, Lamparella N,
Abendroth C and Drabick J: Fatal malignant metastastic epithelioid
angiomyolipoma presenting in a young woman: Case report and review
of the literature. Rare Tumors. 5:e462013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Allolio B and Fassnacht M: Clinical
review: Adrenocortical carcinoma: Clinical update. J Clin
Endocrinol Metab. 91:2027–2037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertherat J and Bertagna X: Pathogenesis
of adrenocortical cancer. Best Pract Res Clin Endocrinol Metab.
23:261–271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lafemina J and Brennan MF: Adrenocortical
carcinoma: Past, present and future. J Surg Oncol. 106:586–594.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fassnacht M, Johanssen S, Quinkler M,
Bucsky P, Willenberg HS, Beuschlein F, Terzolo M, Mueller HH,
Hahner S and Allolio B: German Adrenocortical Carcinoma Registry
Group; European Network for the Study of Adrenal Tumors: Limited
prognostic value of the 2004 International Union Against Cancer
staging classification for adrenocortical carcinoma: Proposal for a
Revised TNM Classification. Cancer. 115:243–250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aubert S, Wacrenier A, Leroy X, Devos P,
Carnaille B, Proye C, Wemeau JL, LecomteHoucke M and Leteurtre E:
Weiss system revisited: a clinicopathologic and immunohistochemical
study of 49 adrenocortical tumors. Am J Surg Pathol. 26:1612–1619.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fassnacht M and Allolio B: Clinical
management of adrenocortical carcinoma. Best Pract Res Clin
Endocrinol Metab. 23:273–289. 2009. View Article : Google Scholar : PubMed/NCBI
|