Introduction
Tumor metabolic reprogramming refers to the
establishment of an entirely new metabolic network under the
abnormal expression of oncogenes and tumor-suppressor genes, by
which the flow of nutrients and energy in the metabolic network is
redefined during the process of tumorigenesis. This reprogramming
results in increased glucose uptake (1,2),
accumulation of lactate (3),
enhanced nucleic acid synthesis (4), lipid metabolism disorders (5) and other alterations (6). Metabolism operates at a higher level
in tumor cells than in normal cells, which is an adaptive process
required to meet the needs of proliferation and migration. Cellular
metabolism and transformation to metastatic disease serve important
roles in tumor development (7–10).
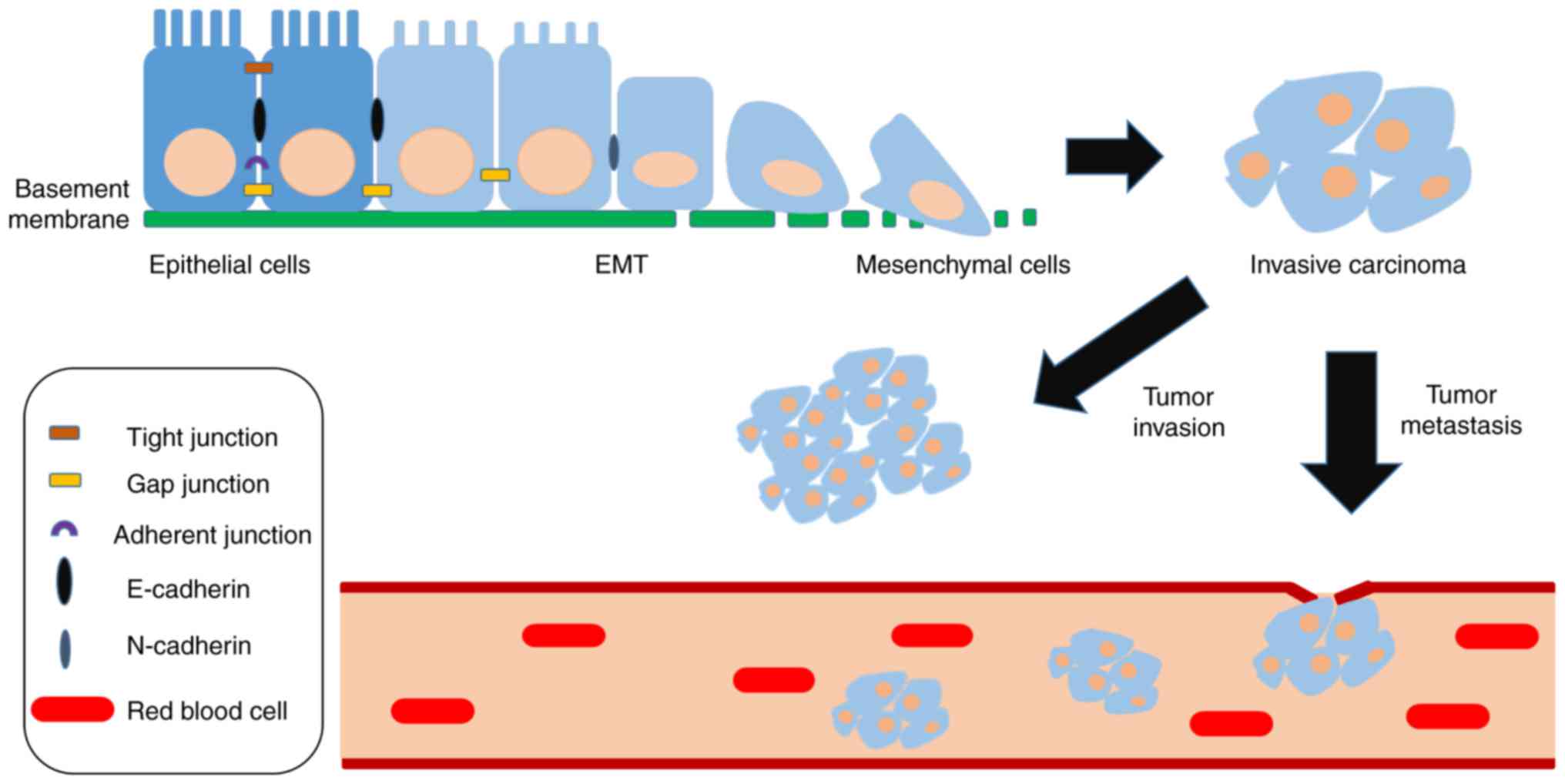
Tumor metastasis is a unique feature of tumor cells
that affects the survival and prognosis of patients with cancer; it
is also an important reason why surgery cannot completely remove
tumor lesions. The epithelial-mesenchymal transition (EMT) is an
important process that occurs prior to tumor metastasis (11–14).
During EMT, tumor cells change from an epithelioid morphology to
mesenchymal cell morphology, with increases in cellular metabolism
and decreases in adhesion between cells; the latter promotes
migration (15–17). Previous studies regarding the
molecular biology of tumor EMT have reported that gene mutations,
deletions and translocations have an impact on various signaling
pathways within cells, including the transforming growth factor
(TGF)-β (18,19), Wnt/β-catenin (20,21),
Notch (22), Hedgehog (23,24)
and interleukin-6/signal transducer and activator of transcription
3 (STAT3) signaling pathways (25,26).
The major carcinogenic signaling pathways ultimately influence
tumor cell metabolism, and such metabolic alterations are essential
for tumor occurrence and development (27). Metabolic reprogramming can provide
both material for cell expansion and sustained signals for
proliferation in order to meet the survival needs of tumor cells in
specific microenvironments (28).
The present study reviewed the metabolic reprogramming that occurs
in EMT, in order to provide novel information that may improve
cancer cure rates.
The EMT process and cancer progression
EMT
Epithelial cells have long been considered to be
terminally differentiated cells that serve protective, supportive
and secretory roles in animals. These cells have a typical
apical-basal polarity. In addition, the presence of intercellular
tight connections and adhesive connections allows epithelial cells
to function together, while also limiting their ability to migrate
freely. Previous studies have reported that the characteristics of
cell polarity, and tight and adhesive connections, can be lost in
epithelial cells (11,29); cells in which these characteristics
are lost are able to infiltrate and migrate, eventually adopting
the morphology and characteristics of mesenchymal cells. This
alteration is defined as the transition from epithelial cells to
mesenchymal cells (i.e., EMT). An early-detected cytokine that
induces EMT was initially named a ‘scatter factor’ and was later
identified as hepatocyte growth factor (HGF); other cytokines that
induce EMT include epidermal growth factor (EGF), fibroblast growth
factor (FGF) and TGF-β (30,31).
Among these, the most common is TGF-β, and it has been confirmed
that TGF-β induces EMT in the majority of epithelial cells and
epithelial-derived tumor cells in vivo and in vitro
(18). Factors affecting the tumor
internal environment, such as hypoxia, also induce EMT (32). The features of EMT are illustrated
in Fig. 1.
 | Figure 1.General characteristics of EMT. The
transition of epithelial cells to a mesenchymal phenotype, induced
by the tumor microenvironment or certain variations, is mainly
manifested in the loss of cell polarity, as well as tight junctions
and adherent junctions, thus resulting in the generation of
mesenchymal cells from epithelial cells, and increased migratory
and invasive capacities. The loss of E-cadherin induced by
upregulated expression of mesenchymal markers (e.g., Snail1/2,
Twist, zinc finger E-box binding homeobox 1/2) is the most
important landmark change associated with EMT, which is often
accompanied by enhanced N-cadherin expression. Compared with
E-cadherin, an increase in N-cadherin is positively correlated with
malignancy, infiltration and metastasis of cancer cells, and
directly affects patient prognosis. EMT, epithelial-mesenchymal
transition. |
E-cadherin reduction or loss
The reduction or loss of E-cadherin, a type I
cadherin, is the most important landmark change associated with
EMT. In cell adhesion, the extracellular domain of E-cadherin binds
to that of E-cadherin molecules in adjacent cells, whereas the
intracellular region forms a complex with α-catenin, β-catenin and
actin (33). In addition to
mediating a strong physical connection between cells, E-cadherin is
an important molecule that maintains epithelial cell identity.
Cells that lose E-cadherin expression are characterized as
non-epithelial cells and exhibit loss of polarity. Upon decreases
in the levels of E-cadherin, intracellular β-catenin is released
and translocates to the nucleus, where it forms a β-catenin/LEF
complex that activates specific mesenchymal gene transcription. A
relationship between E-cadherin and epithelial cancer has
previously been reported (34).
Furthermore, mutations in the E-cadherin gene, or downregulation of
E-cadherin protein expression, are detected in lung, breast,
gastric and prostate cancer, as well as other types of epithelial
cancer (34). The E-cadherin gene
is considered a novel tumor-suppressor gene, mutation and loss of
which are key molecular events during cancer progression and
metastasis (34).
Loss and relocalization of zonula
occludens-1 (ZO-1)
Tight junctions, which are located just below the
apical surface of epithelial cells, have an important role in
sealing gaps between cells. One of the molecules involved is ZO-1,
which binds to the cytoskeleton and acts as a signaling molecule in
the cell. Following EMT, downregulation of ZO-1 protein expression
and interruption of tight junctions are observed (35). In addition, ZO-1 at the membrane can
migrate to the cytoplasm and even the nucleus. Nuclear ZO-1
activates the EGF receptor, promotes the proliferation of cancer
cells and augments the infiltration capacity of pancreatic tumor
cells (35). The N-terminal of the
ZO-1 protein is critical for targeting cell borders, and N-terminal
mutants result in a marked alteration in cell shape and patterns of
gene expression associated with the mesenchymal phenotype in
corneal epithelial cells (36).
EMT and the extracellular matrix
A tumor is a complex cell cluster, the growth and
infiltration of which are associated with the microenvironment
formed by the extracellular matrix. A previous study reported that
extracellular matrix components can promote the occurrence of EMT
in tumor cells, and that cells undergoing EMT can alter the
composition of the extracellular matrix to facilitate tumor
infiltration and metastasis. For example, some tumor cells secrete
matrix metalloproteinase (MMP)3 and MMP28 into the extracellular
matrix, inducing EMT via Ras-related C3 botulinum toxin substrate 1
and reactive oxygen free radicals (37). In primary hepatocellular carcinoma,
EMT can promote expression of MMP1, MMP2 and MMP7 in cells via
upregulation of Snail and zinc finger E-box binding homeobox (ZEB2)
transcription factors, thus resulting in the degradation of
extracellular matrix proteins and enhancement of tumor cell
infiltration (38).
Growth factor-induced EMT
EMT can be induced during tumor progression
(39). Growth factor signaling
pathways lead to loss of E-cadherin function through protein
degradation and gene mutation. In several tumors, the
tumor-associated stroma produces various growth factors and
cytokines, including HGF, EGF, platelet-derived growth factor,
FGF2, tumor necrosis factor (TNF)α, insulin-like growth factor and
TGF-β. TGFβ-induced EMT is the most prominent type of EMT, which
can be regulated by lipogenesis-related energy production (40). All of the aforementioned growth
factors and cytokines may induce the expression of various
E-cadherin transcription inhibitors, including Snail1/2, ZEB1/2 and
Twist (34,41), thus inhibiting the expression of
E-cadherin, eventually resulting in EMT.
EMT also relies on the activation of various
signaling pathways, including the mitogen-activated protein kinase
(MAPK), phosphoinositide 3-kinase (PI3K), Wnt/β-catenin, nuclear
factor (NF)-κB, Notch and Hippo/Warts signaling pathways (34). For example, HGF stimulates EMT
through activation of early growth response-1 via the MAPK
signaling pathway, followed by Snail-1 expression and
downregulation of E-cadherin gene expression (34). Similarly, FGF induces EMT via the
activation of MAPK and TGF-β signaling (34). In addition, Notch signaling can
induce EMT through transcriptional activation of Snail-1 and
Snail-2 through the cooperation with hypoxia (22,42).
Transcription factor-induced EMT
Transcription factors have an important role in EMT
induction, including Snail, Twist and ZEB (43). They can regulate the expression of
certain genes, including E-cadherin, β-catenin, N-cadherin,
vimentin and fibronectin, to suppress substances associated with
the tight linkage of cells. In addition, they are regulated by
intracellular and extracellular signaling pathways to participate
in EMT and promote tumor metastasis.
Snail was the first transcription factor reported to
induce EMT. It can be combined with lysine-specific demethylase 1,
G9a-DNA methyltransferase (DNMT), SUV39H1, mSin3A, histone
deacetylase 1/2 and other enzymes to methylate or acetylate the
histone in the promoter region of E-cadherin, thereby inhibiting
transcription of the target gene (44). In the human skin cancer cell line
A431-III, RNA interference of Snail gene expression, significantly
reduces the secretion of MMP9 and blocks the EMT, thus
significantly reducing tumor invasion and metastasis; conversely,
overexpression of Snail promotes vimentin and N-cadherin
expression, decreases E-cadherin and significantly increases the
secretion of MMP9, thus promoting tumor invasion and metastasis
(45). Haraguchi et al used
ovarian cancer cells (RMZ-1), in which Snail was knocked out by
CRISPR/Cas9 technology, to further clarify the effects of Snail on
the maintenance of cytoskeletal structure, cell migration and
cell-cell adhesion (46). Kim et
al reported that Snail can reprogram glucose metabolism by
suppressing phosphofructokinase, platelet, a major isoform of
cancer-specific phosphofructokinase-1, an enzyme involved in the
first rate-limiting step of glycolysis, thus allowing cancer cell
survival under metabolic stress (47).
Twist is a member of the basic helix-loop-helix
transcription factor family, which includes Twist1 and Twist2.
Twist is also a key regulatory factor involved in EMT and
metastasis. Breast cancer cell lines and tumor animal models have
confirmed that Twist1 can induce EMT and promote tumor invasion and
metastasis; Twist1 acts on the promoter H4K20me1 of E-cadherin and
N-cadherin genes to inhibit E-cadherin gene transcription and
enhance N-cadherin gene transcription through the recruitment of
SET8 (48). When normal breast
epithelial cells and breast cancer cell lines express Twist2, it
can activate the STAT3 signaling pathway to reduce E-cadherin,
resulting in cells obtaining the mesenchymal cell phenotype and
promotion of tumor metastasis. Meanwhile, normal breast epithelial
cells and breast cancer cells obtain a tumor stem cell-like
phenotype through the activation of Twist2 (49).
Overexpression of ZEB protein is closely associated
with tumorigenesis and metastasis. At present, it has been reported
that overexpression of ZEB exists in ovarian, breast, lung,
prostate, colon and bladder cancer (50). ZEB can induce EMT in tumor cells via
the induction of MMPs (50). Gene
chips have been used to detect gene expression in 38 patients with
non-small cell lung cancer; the results revealed that ZEB1 and ZEB2
can regulate the expression of >400 genes, most of which are
closely associated with tumor EMT, including epithelial cellular
adhesion molecule, CDP-diacylglycerol synthase 1, suppression of
tumorigenicity 14, FGF receptor 1 and vimentin (51).
Metabolism and EMT
Metabolic phenotypes of cancer
cells
In early tumor stages, the metabolic phenotypes of
tumor cells are similar to normal cells (1,2). With
the development of EMT, differentiated epithelial cancer cells are
replaced with undifferentiated mesenchymal cancer cells; the
metabolic phenotypes of these cells are also altered (52). To date, the key characteristics of
cell metabolism in mesenchymal cells have been fully addressed.
Increased glycolytic flux
Aerobic glycolysis is increased in mesenchymal
cancer cells to minimize reactive oxygen species (ROS) (53), which are the main source of cellular
metabolic damage partially produced by oxidative phosphorylation.
Aerobic glycolysis better satisfies the basic needs of dividing
cells: Rapid ATP production, increased macromolecular biosynthesis
and enhanced maintenance of appropriate cellular redox status
(54). During EMT, glycolysis is
more active in cancer cells, which not only satisfies the basic
needs of cancer cells for appropriate energy levels, adequate
biosynthetic precursors and balanced redox status, but also helps
maintain a poorly differentiated state (55).
Rapid ATP generation from
glycolysis
Glucose metabolism provides ATP to cells through
mitochondrial oxidative phosphorylation and glycolysis. However,
since the ATP provided by oxidative phosphorylation in mesenchymal
cancer cells usually does not meet the proliferation requirements,
cells tend to use glycolysis, a pathway with low glucose
utilization but rapid delivery of ATP, to obtain energy (56,57).
Increased pentose phosphate pathway
(PPP) flux
Alterations in the PPP are directly associated with
the growth state of cells. The microenvironment of undifferentiated
mesenchymal cancer cells serves a key role in the metabolic
phenotype of cancer cells. Hypoxia and lactic acid accumulation
promote cancer cells to increase the flow of PPP, providing a
material basis for the rapid growth of cancer cells (58). Two aspects determine the redox
equilibrium state of cells: The production and elimination rate of
ROS. It has been reported that ROS links glucose metabolism to the
EMT phenotype in basal-like breast cancer. and that the EMT
phenotype promotes metabolic conversion to glucose metabolism,
leading to decreased ROS levels (59–61).
The loss of fructose-bisphosphatase 1, a rate-limiting metabolic
enzyme of gluconeogenesis, can inhibit oxygen consumption and ROS
production by suppressing the activity of mitochondrial complex I
and increasing the synthesis of NADPH through PPP flux, which
subsequently increases the mesenchymal phenotype in breast cancer
(55).
Activated Thr-Gly metabolism
Threonine and glycine can synthesize S-adenosyl
methionine (SAM), which is a common substrate for DNA and histone
methylation in cells, by folate metabolism (62). DNMTs and histone methyltransferases
(HMTs) can transfer methyl groups from SAM to substrates, in order
to form the by-product S-adenosyl homocysteine (SAH), which is an
effective inhibitor of DNMT and HMTs, thereby adding methyl groups
to DNA or lysine/arginine residue of histones (63). In mesenchymal cells, Thr-Gly
metabolism is activated, and threonine dehydrogenase promotes SAM
synthesis while converting threonine to glycine and acetyl-CoA,
resulting in a high ratio of SAM/SAH. SAM and SAH hydrolase can
both activate enhancer of zeste 2 polycomb repressive complex 2
subunit, a methyltransferase involved in suppressing gene
expression through methylation of histone H3 on lysine 27,
resulting in H3K27me3-dependent inactivation of E-cadherin
(64).
Glucose metabolism during EMT
Glucose metabolism
Compared with normal cells, tumor cells exhibit a
stronger ability to absorb glucose; however, the capacity to use
this substance is weak, even under conditions of sufficient oxygen.
As tumor cells primarily invoke glycolysis to obtain energy, which
is accompanied by lactic acid accumulation and a small amount of
ATP, an anoxic environment is generated. This process is called the
Warburg effect (56,57); compared with primary tumors, the
Warburg effect is more prominent in metastatic tumors.
Mitochondrial dysfunction
Mitochondria are the main organelles that supply
energy in human cells, and are also involved in the transmission of
apoptotic signals in normal cells. These functions of mitochondria
determine their close association with numerous diseases, including
cancer. It has been reported that mitochondrial dysfunction is
significantly associated with glucose metabolic reprogramming and
the ability to lose autonomic apoptosis in several types of
metastatic cancer (65). The
structure and function of mitochondria in cancer cells is altered,
alongside mitochondrial expansion, increased glucose intake and
lactate accumulation, during EMT, whereas the glucose oxidation
capacity of mitochondria declines (66). In breast cancer and melanoma cells,
the reduced glucose oxidation in mitochondria can enhance the EMT
process to accelerate tumor metastasis (67). Pathological alterations in
mitochondrial membrane permeability can release some apoptotic
proteins; however, the numerous anti-apoptotic signals, including
B-cell lymphoma 2 (Bcl-2) and Bcl-extra large, in tumor cells
inhibit these apoptotic proteins. It has been reported that the
Bcl-2 family member Mcl-1 is expressed during tumor metastasis, and
its anti-apoptotic ability is required for tumor metastasis
(68). With regards to
mitochondrial function in tumor-associated changes, maintaining
normal mitochondrial signaling, enhancing mitochondrial oxidative
capacity, and regulating the expression of apoptotic proteins are
potential directions for clinical research.
Lactic acid accumulation
Glucose metabolism provides ATP to cells through
mitochondrial oxidative phosphorylation and glycolysis; however,
since the supply of ATP due to mitochondrial oxidative
phosphorylation does not typically meet the proliferation
requirements of tumor cells, tumors tend to employ glycolysis to
obtain energy (69,70). A direct result is an increase in the
lactate content of tumor cells; notably, the levels of lactic acid
in tumor cells are directly associated with the EMT process and
metastatic behavior of tumors (71). Accumulation of lactic acid can
induce expression and activation of enzymes associated with
glycolysis, including hexokinase and 6-phosphofructokinase 1, to
enhance the supply of ATP in tumor cells. Tumor cells can use
lactic acid as a source of energy for metabolism and also procure
lactic acid from surrounding cells to maintain the acidic
environment, in order to avoid apoptosis and promote metastasis
(72). Glucose metabolism in
endothelial cells produces lactic acid, which can promote
angiogenesis and provide a suitable environment for metastasis. Liu
et al revealed that EMT induction is associated with
augmented glucose uptake and lactate production in pancreatic
ductal adenocarcinoma (73).
Exogenous lactic acid can also favor metastasis of cancer cells in
a concentration-dependent manner in head and neck carcinoma cells
(74), and these advantages enable
EMT, tumor invasion and metastasis. Furthermore, previous studies
have reported that the lactic acid accumulated in tumor cells
functions through the TGF-β2 signaling pathway to induce tumor cell
metastasis (70,74), and experiments in mice have
demonstrated that cell metastasis is blocked when the glycolytic
pathway is inhibited (75). These
findings indicate that a reduction in lactic acid alters the acidic
environment of tumor cells, eliminates the advantages of the tumor
cell microenvironment, and reduces available energy sources for
tumor cell metabolism; this may be a potential strategy to prevent
tumor cell metastasis.
Pyruvate
During glycolysis, phosphoenolpyruvate, the
intermediate that is converted to pyruvate by pyruvate kinase, is
key. It has been reported that normal cells mainly express pyruvate
kinase muscle (PKM)1, whereas tumor cells aberrantly express PKM2
(76). In addition, it has been
demonstrated that c-Myc gene-mediated selective cleavage is the
main reason for tumor-specific expression of PKM2 (77), which is mainly present in the form
of a dimer. PKM2 promotes the biosynthesis of glycolytic
intermediates. In addition, PKM2 dimers participate in signal
transduction in the form of active phosphokinase and are involved
in the regulation of gene transcription together with β-catenin
(78,79), hypoxia-inducible factor (HIF) and
other transcription factors following nuclear translocation
(80). The nuclear translocation of
PKM2 induced by stroma impairs oxidative phosphorylation and
metastatic spread in prostate cancer (81). Abnormal expression of PKM2 is
positively correlated with EMT in esophageal squamous cell,
gallbladder and papillary thyroid cancers (82–84),
and interference of PKM2 expression significantly inhibits tumor
growth, invasion and metastasis (85).
HIF
Tumor cells grow rapidly in the body, and rapid
proliferation, vascular remodeling and a lack of blood supply,
generally mean that these cells are maintained in an anoxic state.
A hypoxic microenvironment can enhance the resistance of tumor
cells to chemotherapy, reducing the survival rate of patients. This
state can also activate HIF-1α, which is associated with tumor
metabolism and metastasis-associated gene activation (86–88). A
previous study reported that HIF-1 may promote EMT and metastasis
(89). Song et al reported
that HIF-1α can increase the gene expression levels of pyruvate
kinase-2, phosphokinase-1, glucose transporter (GLUT)1 and lactate
dehydrogenase A (LDHA) expression under hypoxic conditions,
promoting carbohydrate metabolism in cells, and providing energy to
promote tumor growth and metastasis during EMT (90). In addition, instantaneous activation
of HIF-1α can induce LDHA expression and E1 subunit phosphorylation
of pyruvate dehydrogenase, resulting in the transition from
intracellular glucose metabolic pathways and mitochondrial
oxidative phosphorylation to anaerobic glycolysis and lactate
fermentation (91). When this
transition is inhibited by an HIF-1α inhibitor,
3-(5′-hydroxymethyl-2′-furan)-1-benzylindazole, the levels of ROS
produced during chemotherapy are increased, and the formation of
metastatic lung tumors is inhibited (92,93).
Therefore, reducing HIF-1α expression in tumor cells and decreasing
the enhancement of glycolysis by HIF-α in tumor cells may reduce
their energy intake, inhibit EMT and reduce metastasis.
HIF-1α can activate TGF-β/Smad3 and Wnt signaling
pathways, upregulate the expression levels of TWIST and Snail, and
promote the invasion and metastasis of tumor cells through EMT
induction. It has previously been reported that the expression
levels of HIF-1α and EMT-associated transcription factors are
positively associated in tumor cells (64). HIF-1α can directly or indirectly
induce EMT-associated gene expression, thus promoting EMT in breast
cancer (52). Previous studies have
revealed that HIF-1α regulates >40 factors, including Snail,
ZEB2, TWIST, Slug, carbonic anhydrase IX, etc., through various
signaling pathways to support tumor metastasis (78,79).
Glucose metabolism and drug
resistance
In the majority of cancers, drug resistance is the
leading cause of chemotherapy failure. It has been reported that
cellular metabolic disorders are associated with this resistance.
Ippolito et al revealed that docetaxel-resistant PC3
(PC3-DR) cells undergo EMT and possess strong metastatic properties
(94). Furthermore, PC3-DR cells
have a more efficient metabolic phenotype than other cells,
involving the use of glucose, glutamine and lactic acid by
mitochondrial oxidative phosphorylation; therefore, compromising
this metabolic reprogramming may be a successful treatment
strategy.
Lipid metabolism during EMT
Fatty acid metabolism and EMT
Fatty acids are essential constituents of biofilm
lipids and signaling molecules, and are important substrates for
energy metabolism (95,96). When cultured in the same medium,
lipids in adipocytes can be redirected to ovarian cancer cells to
provide energy for expansion of cancer cells (97). However, tumor cells do not ingest
exogenous fatty acids in vivo but rather consume a large
amount of ATP and NADPH (to synthesize one fatty acid molecule, 14
molecules of ATP and seven molecules of NADPH are required), a
process termed fatty acid de novo synthesis. The fatty acids
required for malignant tumor growth and proliferation are mainly
derived from the de novo synthesis pathway, and fatty acid
de novo synthesis is a unique characteristic of malignant
cancer (5). Menendez et al
suggested that tumor cells are always self-synthesizing fatty acids
to maintain rapid proliferation and achieve survival (98). In addition, the fatty acid synthesis
pathway protects cells from oxygen free radicals and
chemotherapeutic agents by consuming reduced equivalents of NAPDH
to balance cell oxidation-reduction levels. These conditions
provide the energy and environment for tumor EMT and ultimately
promote tumor metastasis.
Fatty acid synthase (FASN) and
EMT
FASN, the only key enzyme in fatty acid de
novo synthesis, is expressed in tumors, such as breast cancer
and melanoma (99). Due to the
increased levels of FASN expression, >90% of triglycerides are
synthesized de novo, despite the presence of high levels of
free fatty acids in cells. Tumor cells are highly dependent on the
de novo synthesis of fatty acids, such that inhibition of
fatty acid synthesis through FASN will selectively induce apoptosis
in human cancer cells, but has little effect on normal cells
(100). FASN serves a crucial role
in tumor EMT and regulates the process in two ways: i) FASN induces
the composition and stability of lipid rafts, which in turn affect
proteins located in the rafts to promote phosphorylation of
E-cadherin and lead to its degradation, weakening intercellular
contact and inducing EMT (101–103); ii) FASN induces the occurrence and
progression of EMT by altering the palmitoylation level of Wnt and
activating the Wnt/β-catenin signaling pathway (102). Inhibition of FASN can reverse EMT
in breast cancer and colorectal cancer cells, thus inhibiting tumor
cell invasion, metastasis and drug resistance (102,103); therefore, the study of FASN
inhibitors may provide a novel means for tumor treatment and
prognosis.
Cholesterol and EMT
Cholesterol is a key component of the cell membrane,
particularly the cytoplasmic membrane (104), and is a precursor of such
compounds as steroid hormones, bile acids and vitamins (105). Previous studies have revealed that
cholesterol metabolism is closely associated with tumor EMT
(106,107). For example, Alikhani et al
demonstrated that, in a breast cancer animal model, abnormal levels
of plasma cholesterol promote EMT and accelerate tumor cell
metastasis (108). Furthermore, it
has been reported that the use of statins to inhibit cholesterol
synthesis can suppress cancer invasion and metastasis (109–112). The specific molecular mechanism by
which cholesterol metabolism affects tumor EMT is not yet clear;
however, there are numerous hypotheses that may be involved.
Firstly, a lipid raft is a microstructure in the
plasma membrane that is rich in cholesterol and sphingolipid, and
serves an important role in biological processes, including signal
transduction and cytoplasmic membrane protein sorting (113). As a key component of lipid rafts,
alterations in cholesterol levels can affect the structure and
function of lipid rafts (114).
Lipid rafts are closely associated with signal transduction in EMT
through such factors as osteopontin (OPN), which is an important
tumor-promoting molecule. OPN activates MAPK, PI3K/AKT
serine/threonine kinase (AKT) and NF-κB, through its receptor
protein αvβ3 integrin and cluster of differentiation (CD)44, which
are located on lipid rafts (115,116), in order to enhance the expression
of MMP2, MMP9, urinary plasminogen activator and other genes, thus
promoting tumor EMT (117).
Furthermore, reducing cholesterol levels in the plasma membrane to
destroy lipid raft structure can cause removal of CD44 from the
membrane, thus inhibiting EMT in tumor cells (118). In addition, lipid rafts have an
important role in activation of the EGF receptor (EGFR) signaling
pathway, which is an important signaling pathway in tumor invasion
and metastasis (119). It has been
reported that breast cancer cell lines exhibit resistance to EGFR
tyrosine kinase inhibitors when EGFR is located in lipid rafts, but
using methyl-β-cyclodextrin or statins to reduce cholesterol and
destroy lipid rafts can counteract this effect (120).
Secondly, farnesyl pyrophosphate (FPP) and
geranylgeranyl pyrophosphate (GGPP) are two important metabolites
in the bypass route of cholesterol metabolism. These two
metabolites are involved in the prenylation of Ras and Rho
proteins; prenylation is necessary for the binding and activation
between Ras and Rho and the cell membrane (112). A previous study regarding glioma
reported that inhibition of cholesterol synthesis leads to a
decrease in FPP and GGPP metabolites, thus inhibiting the
Ras-Raf-MAPK kinase-extracellular signal-regulated kinase (ERK)
signaling pathway and leading to a decrease in the growth and
invasion of glioma cells (121).
Finally, research on breast cancer has revealed that
abnormal cholesterol metabolism causes accumulation of its
metabolite 27-hydroxycholesterol, which promotes breast cancer
growth and EMT by activating estrogen receptor (ER) and liver X
receptor (122,123).
Amino acid metabolism during EMT
Amino acids in tumor cells
Tumor cells not only require the necessary energy
reserves to maintain their survival, but also a large amount of
biological raw materials that are used to generate new sub-cells.
Previous studies have reported the significance of abnormal
metabolism of glutamate, aspartic acid, glycine and serine, as well
as other amino acids, during tumor growth, invasion and metastasis
(70,124,125). The present review described the
mechanisms by which these amino acids are metabolically broken down
during tumor metastasis.
Glutamine and EMT
Roberts and Frankel, and Roberts and Borges reported
that some tumor types predominantly use glutamine for survival
(126,127). These tumor cells do not
necessarily rely on glucose uptake but rather on
glutamine-dependent growth, a phenomenon called ‘glutamine
addiction’ (128,129). Glutamine predominantly produces
α-ketoglutarate (α-KG) in the mitochondria and provides the raw
material for oxidative phosphorylation and lipid synthesis through
the tricarboxylic acid cycle (TCA) cycle (70). In addition, glutaminase (GLS) and
glutamate dehydrogenase 1 (GDH1), which are the key enzymes of
glutamine metabolism, are highly expressed in lymphoma and prostate
cancer cells and are closely associated with the classification and
prognosis of a tumor via regulation of the oncogene c-Myc (130). Reducing expression of GLS or GDH1
can inhibit lipid synthesis or induce intracellular redox stress,
thus resulting in apoptosis of tumor cells (131,132). Furthermore, oxidative stress
during tumor progression is an important factor for inhibiting the
distant metastasis of melanoma cells (133). Conversely, fumarate, which is the
intermediate metabolite of glutamine metabolism, can reduce the
levels of ROS in breast cancer cells by activating glutathione
peroxidase, thus maintaining the redox balance (95,132),
which may promote the EMT process and metastasis.
Additionally, in the human body, different types of
cells use distinct metabolic pathways due to their phenotypic
differences. In breast cancer, basal-like cancer cells with the
mesenchymal phenotype are more sensitive to glutamine-targeted
therapy than luminal cells with an epithelial phenotype, possibly
due to different patterns of glutamine metabolism (134). However, the specific mechanism
underlying abnormal amino acid metabolism in tumors remains to be
elucidated.
Glycine and EMT
Glycine has an important role as a provider of
carbon units. Analysis of the in vivo metabolic pathway in
patients with glioma revealed that glucose ingested by tumor cells
can be used as a carbon source for de novo synthesis of
glycine (135) (Fig. 2). This experiment revealed that
glycine de novo synthesis has a very important role in the
progression of human tumors. In addition, metabolic analysis of
NCI-60 tumor cell lines indicated that the rapid proliferation of
tumor cells is increasingly dependent on glycine, and that the
proliferation rate of tumor cells has a close association with
glycine consumption and the expression levels of molecules that are
important for mitochondrial glycine synthesis (136). High levels of glycine synthesis
are a prerequisite for tumor EMT and also contribute to tumor
metastasis in lung cancer (136).
Glycine is also a building block for protein synthesis, and isotope
labeling revealed that glycine is utilized in the synthesis of
purines, which are key raw materials for nucleic acid synthesis,
and proteins (Fig. 2) (137). Expression of glycine decarboxylase
(GLDC), which is a rate-limiting enzyme of the glycine cleavage
system, is abnormally high in tumor-initiating cells of non-small
cell lung cancer (138). GLDC
functions in a manner similar to that of oncogenes, and
overexpression of GLDC alters metabolism of glycine, glycolysis and
synthesis of pyrimidines, which are also an important component of
nucleotides (Fig. 2). In addition,
glycine is a glycogenic amino acid and can be converted by serine
transhydroxymethylase to serine, from which pyruvate is then
generated (139). Therefore,
glycine metabolism is an important pathway for serine biosynthesis.
Conversely, it has also been reported that the addition of glycine
to food inhibits proliferation of melanoma B16 cells in mice
(140); the mechanism underlying
these effects is considered to occur via food-borne glycine
inhibition of vascular endothelial cell proliferation, thus leading
to disordered angiogenesis.
 | Figure 2.Schematic diagram of the metabolic
reprogramming of cancer cells during EMT. Various aspects of
metabolic reprogramming during EMT are shown, including glycolysis,
the TCA cycle, pentose phosphate pathway, glutaminolysis, and
biosynthesis of fatty acid, nucleotide and amino acid pathways. The
key molecules, including c-Myc, p53, HIF-1α, RAS and AKT,
coordinate regulation of cancer metabolism in different ways. ACL,
ATP-citrate lyase; α-KG, α-ketoglutarate; AKT, AKT serine/threonine
kinase; F6P, fructose-6-phosphate; FAS, fatty acid synthase; G6P,
glucose-6-phosphate; G6PDH, glucose-6-phosphate dehydrogenase;
GLDC, glycine decarboxylase; GLS, glutaminase; Glut1, glucose
transporter 1; HIF, hypoxia-inducible factor; LDH, lactate
dehydrogenase; MCT, monocarboxylate transporter, OXPHOS, oxidative
phosphorylation; PDH, pyruvate dehydrogenase; PFK,
phosphofructokinase; PHGDH, phosphoglycerate dehydrogenase; PKM2,
pyruvate kinase muscle 2; SCO2, synthesis of cytochrome c
oxidase 2; TCA, tricarboxylic acid. |
Serine and EMT
Although serine is a non-essential amino acid, it
has an indispensable role in metabolism. Serine is involved in the
biosynthesis of S-adenosylmethionine and is the most important
carbon unit in the methylation reaction (141). Phosphoglycerate dehydrogenase
(PHGDH) catalyzes the first step of serine synthesis and can also
control the flow of glycolytic intermediates into the serine
biosynthetic pathway (142)
(Fig. 2). The PHGDH gene is located
in the region of recurrent copy number gain in breast cancer, and
expression of the PHGDH protein is upregulated in 70% of
ER-negative breast cancer tissues (142). The serine synthesis flux in breast
cancer cells with high PHGDH protein expression is increased, and
inhibition of PHGDH expression in cells with high PHGDH protein
expression can reduce serine synthesis and decrease the ability of
cells to proliferate. Serine is a glucogenic amino acid that can be
converted to pyruvate, α-KG and other TCA cycle intermediates, in
order to participate in gluconeogenesis, thus generating glucose.
However, a decrease in PHGDH does not result in a decrease in
intracellular serine concentration but induces a decrease in α-KG,
which can be converted to glutamine by glutamine synthase via the
mitochondrial TCA cycle (142). It
has previously been reported that ~50% of the glutamine flow
entering the TCA cycle is from the serine synthesis pathway in
cells with high PHGDH protein expression (142). Serine can also be decomposed into
acetyl-CoA, which is a main participant in the TCA cycle, and can
enter TCA cycle oxidation; acetyl-CoA can be further converted to
fatty acids by FAS. Therefore, it is reasonable to speculate that
the synthesis of serine in tumor cells can be used for fatty acid
synthesis. Abnormal expression of serine and its related enzymes in
tumor cells is likely to be an important factor in the metabolic
reprogramming of tumor cells, as well as a source of energy for
tumor development. Inhibition or reversal of these abnormal
alterations may provide novel ideas for inhibiting tumor
progression, including the development of EMT.
Nucleic acid metabolism during EMT
Nucleic acids are among the most basic molecules,
which serve a decisive role in such processes as growth,
inheritance and variation. Cells in different organisms have
particular characteristics; in addition, the metabolism of tumor
cells and normal cells in the human body differs significantly, and
the various levels of development of tumor cells also differ
widely. It has been reported that thymidylate kinase and thymidine
kinase are key for de novo synthesis and salvage synthesis
of pyrimidine nucleotides, and are more active in tumor cells than
in normal cells (4). This
phenomenon is more pronounced during tumor metastasis. With the
development of molecular imaging technology, alterations in cell
morphology can be observed and detection of nucleic acid synthesis
during tumor cell metastasis can be conducted (143). During metastasis, the rapid
proliferation of cells includes the transmission of genetic
information, transcription, translation and other process,
inevitably leading to increased synthesis of DNA and RNA. Although
food contains a sufficient amount of nucleic acids, only a small
portion of their decomposition products can be used for
re-synthesis of nucleic acids. Notably, a large portion of the raw
material required for nucleic acid synthesis is derived from
carbohydrate metabolism; for example, ribose 5-phosphate produced
via degradation of glucose 6-phosphate in the PPP can be catalyzed
to pyrophosphate 5-phosphate with ATP, via a specific
ribose-phosphate pyrophosphokinase, for mononucleotide synthesis.
Therefore, carbohydrate metabolism is considered the material basis
of nucleic acid synthesis during tumor metastasis (58). With the development of molecular
biology, the purpose of which is to identify mechanisms, the focus
of research on tumor metastasis is largely conducted at the genetic
level; for example, microRNA (miR)-10b promotes cell metastasis and
the mesenchymal subtype of glioblastoma multiforme via the
suppression of tumor protein p53 (TP53), paired box 6, NOTCH1 and
homeobox D10 (144); long
non-coding RNA HOX transcript antisense RNA induces genome-wide
re-targeting of polycomb repressive complex 2 to an occupancy
pattern more resembling embryonic fibroblasts, leading to altered
histone H3 lysine 27 methylation, gene expression, and increased
cancer invasiveness and metastasis (145). Conversely, little attention has
been given to the metabolism of macromolecules. In tumor metastasis
research, inhibition or activation of certain enzymes at the level
of nucleic acid synthesis may reveal a novel target for tumor
therapy.
Enzymes and EMT
In the process of EMT during tumor metastasis,
enzyme catalysis is highly relevant for nucleic acid, carbohydrate
and lipid metabolism. As an example, glucose-6-phosphate
dehydrogenase, which is a key enzyme in the PPP, is capable of
continuously synthesizing nucleotides and lipids to meet the
material and energy requirements of tumor metastasis. Therefore,
glucose-6-phosphate dehydrogenase is considered a potential
antitumor target (146).
Oxygenated glycosylated proteins can enhance glycosyltransferase
activity and are therefore a major marker of migration in ovarian
cancer cells. Because glycosylation of polypeptide
N-acetylgalactosyltransferase 14 (GALNT14) is associated with
transmembrane mucin-13, and both are found at high levels in
ovarian cancer cells, suppression of the GALNT14 gene by small
interfering RNA may inhibit cell metastasis, indicating that
GALNT14 can promote tumor metastasis by modulating transmembrane
mucin-13 (147). It has been
reported that heparanase serves an important role in tumor invasion
and metastasis, and tumor cell invasion and metastasis are
inhibited by the heparanase inhibitor OGT2115, as well as
low-molecular-weight heparin (148). The reduced form of NADPH oxidase
is the main source of ROS, and ROS can induce apoptosis of
endothelial cells in vivo, which is a prerequisite for tumor
cells to escape from the circulatory system and migrate to other
tissues and organs (149). It has
been reported that angiotensin-converting enzyme-2, which is a key
enzyme in the kidney-angiotensin system, can attenuate the
metastasis of non-small cell lung cancer through inhibition of EMT
(150). Furthermore, AMP-activated
protein kinase (AMPK) induces cell transfer, although its function
remains unclear; knockout of the AMPK gene in an ovarian cancer
xenograft model can significantly inhibit tumor metastasis to the
lungs, whereas AMPK activation induced by lysophosphatidic acid
promotes ovarian cancer cell metastasis (151). However, AMPK-α2 protects against
liver injury from metastasized tumors via reduced glucose
deprivation-induced oxidative stress, including alleviating hepatic
hypoglycemia and mitochondria-mediated inhibition of ROS production
(152).
These results indicate that different enzymes serve
important roles in the process of tumor metastasis through distinct
pathways. By studying the regulation of enzymes during tumor cell
metastasis, the synthesis rate of nucleotides and lipids can be
specifically reduced, as can the energy utilization of tumor cells.
The use of inhibitors to inhibit the activity of
metastasis-associated enzymes is one of the most commonly applied
clinical treatments.
EMT and CSCs
Cancer stem cells (CSCs) are a small population of
cells within tumor tissues, which possess the capacities of
self-renewal, multipotent differentiation, unlimited proliferation
and strong tumorigenic ability (153). They maintain tumor growth and
proliferation. The insensitivity of tumors to radiotherapy and
chemotherapy, tumor recurrence and metastasis are all closely
associated with CSCs. To date, scientists have isolated CSCs in
various solid tumors, such as liver, breast, lung, and brain stem
tumors (154–157).
By comparing the main metabolic patterns of
metastatic prostate epithelial CSCs (e-CSCs) with non-CSCs
expressing stable EMT. Aguilar et al revealed that e-CSCs
possess a high degree of plasticity with regards to energy
metabolism, including enhanced glycolysis, fat and amino acid
metabolism, and glutamine metabolism (158), which indicates that metabolic
reprogramming of cancer cells predominantly occurs in CSCs. Correct
identification of key metabolic participants that are regulated
post-transcriptionally can provide potential biomarkers and
therapeutic targets for the effective prevention of metastasis
(158).
EMT is a key developmental program that is often
activated during cancer invasion and metastasis. It has been
reported that EMT can transform immortalized human mammary
epithelial cells into cells with CSC properties, thus indicating
that EMT-transformed cells have stem cell properties, and inducing
EMT can not only promote the proliferation of tumor cells from the
primary site, but also enhance the self-renewal ability of tumor
cells (159). Therefore, EMT can
transform tumor cells into CSCs, and CSCs may acquire an increased
migratory ability through EMT transformation (160).
The majority of poorly differentiated cancers
usually evade cancer treatment; however, the reason is unclear.
Oshimori et al indicated that TGF-β, which is concentrated
in the tumor vasculature, confers a longer cell renewal cycle of
adjacent squamous cell carcinoma stem cells (SCC-SC). SCC-SC, in
response to TGF-β, exhibits enhanced protection against anticancer
drugs; however, a longer cell renewal cycle alone will not improve
survival. In addition, TGF-β activates p21 and stabilizes nuclear
factor (erythroid-derived 2)-like 2, thereby significantly
enhancing glutathione metabolism and therapeutic resistance
(161). Furthermore, activated
TGF-β can bind to the high-affinity receptors TβRI and TβRII, which
have serine/threonine protein kinase activity, on the tumor cell
membrane to activate Smad2 and Smad3 (19). Smad2 and Smad3 enter into the
nucleus alongside Smad4 to interact with other transcription
factors, inducing the expression of target genes, such as Snail1,
Snail2 and ZEB, eventually leading to reduction of E-cadherin and
induction of N-cadherin and vimentin, thus resulting in increased
motility of tumor cells (19).
TGF-β can also induce EMT through non-Smad pathways by activating
ERK MAPK, Rho GTPases, PI3K/AKT and other signaling pathways
(19).
It has been reported that under hypoxic conditions,
CSCs are more prone to EMT, leading to cell morphological
alterations and enhanced metastatic capacity. However, EMT can be
reduced when the normoxic control level is restored, indicating
that this is a reversible process. Notably, due to the action of
cytokines such as TNFα, even under normoxic conditions, there is
still a small portion of CSCs that undergo EMT (162). These differences in oxygen
metabolism patterns provide an explanation for the general
therapeutic resistance of CSCs and the greater resistance of CSCs
that develop EMT.
Biddle et al reported that non-EMT and EMT
CSCs can switch their epithelial or mesenchymal traits to
reconstitute cellular heterogeneity; this is a characteristic of
CSCs (163). CSCs can switch
between epithelial and mesenchymal cells via EMT and
mesenchymal-epithelial transition. Previous studies revealed that
post-EMT CSCs exhibit particularly enhanced therapeutic resistance
(163,164). In conclusion, selecting an
appropriate targeted therapy based on the different phenotypes of
CSCs is key to effective outcomes.
Metabolism-associated oncogenes and
tumor-suppressor genes
Association of oncogenes and
tumor-suppressor genes with metabolism
Rapid proliferation of cells requires a sufficient
amount of new biological macromolecules for cellular construction.
These biological macromolecules vary widely, and may be obtained
directly from the extracellular environment or by metabolism of
glucose, which is a common nutrient for the synthesis of other
required biological macromolecules. Therefore, the maximum
utilization of available nutrients and conversion into other
molecules through complex metabolic networks is the most important
goal of tumor cell metabolism. Previously, researchers believed
that a tumor is the result of oncogene activation or
tumor-suppressor gene inactivation, and that metabolic
abnormalities were merely concomitant phenomena of the genetic
events that induce tumors (28).
However, the results of cell metabolism studies in recent decades
have revealed that genetic events, including oncogene activation
and tumor-suppressor gene inactivation, thus affecting genes such
as Kras, p53, c-myc and isocitrate dehydrogenase (IDH), may be
initiators of malignant cell transformation, with metabolic
reprogramming being an essential pathway underlying the genetic
events that mediate malignant transformation, including EMT, to
promote tumor metastasis (165–168). Notably, increasing evidence has
suggested that several classical oncogenes or tumor-suppressor
genes are directly or indirectly involved in metabolic
reprogramming (169–171). As aforementioned, tumor metabolism
reprogramming may have an important role in promoting EMT.
Kras
Carcinogenic mutations of the Kras gene are an
early event in pancreatic cancer. The proliferation of mouse
pancreatic cancer driven by the mutant Kras gene Kras(G12D), is
highly dependent on Kras(G12D) expression, resulting in
reprogrammed cellular metabolism to support proliferation (165). Kras(G12D) can stimulate tumor
cells to take up glucose and drive glucose intermediate metabolites
into the hexose amine biosynthesis and the PPP pathways to promote
ribose synthesis. Unlike the classical model, Kras(G12D) drives
glycolysis intermediates into the non-oxidized PPP, resulting in
loss of contact between ribose synthesis and NADP/NADPH-mediated
redox control (165). Kras
metabolizes glucose and glutamine to support cell growth and
promotes growth capacity that is not dependent on docking (172). This effect is achieved by
regulating the ERK/MAPK signaling pathway to induce ROS production
and glutamine entry into the TCA cycle to be metabolized to α-KG
(173) (Fig. 2). In addition, H-ras (G12V) or K-ras
(G12V) can increase the number of functional mitochondria in cells
and maintain mitochondrial metabolism by increasing autophagy to
promote tumor cell survival (174).
p53
As a well-known tumor-suppressor gene, p53 is also
the most important negative regulator during tumor metabolic
reprogramming, which has been reported to regulate EMT and
metastasis in colorectal cancer (166). In addition, p53 inhibits
glycolysis, promotes fatty acid oxidation and is associated with
the maintenance of an appropriate redox status (58). p53 can inhibit expression of GLUT1,
GLUT4 and phosphoglycerate mutase to upregulate expression of
TP5-induced glycolysis regulatory phosphatase, thereby suppressing
glycolysis (175–177). In addition, p53 can promote
expression of synthesis of cytochrome c oxidase 2 (SCO2),
which is critical for regulating the cytochrome c oxidase
(COX) complex (169) (Fig. 2). Decreased p53 expression in cancer
cells results in decreased SCO2 protein expression and reduced
oxygen consumption, which provides a possible explanation for the
Warburg effect, and offers novel information as to how p53 may
affect tumor growth and metastasis. Furthermore, in serous ovarian
cancer cells, a mutated p53 gene can enhance lipid metabolism to
promote tumor cell metastasis, accompanied by epithelial cell
transformation to mesenchymal cells, fatty acid uptake promotion in
adipocytes and invasive alterations in other tumor cells,
eventually leading to decreased patient survival (178).
c-Myc
The well-known tumorigenic transcription factor
c-Myc can induce EMT and metabolic reprogramming through numerous
routes. It can stimulate glycolysis, glutaminolysis and nucleotide
synthesis (135,170); induce EMT in mammary epithelial
cells; cooperate with TGF-β in promoting EMT and metastasis
(179); and promote EMT and
angiogenesis through upregulating miR9 (167). c-Myc-dependent tumor cells appear
to be ‘glutamine-addicted’. c-Myc can also promote the use of
glutamate in tumor cell mitochondria by activating the enzymes
involved in glutamine metabolism, an effect known as glutaminolysis
(134). In addition to directly
regulating expression of metabolic enzymes, c-Myc stimulates the
conversion of glutamine to glutamate in tumor cells, and its entry
into the TCA cycle. This results in generation of ATP to support
proliferation via inhibition of miR-23a/b transcription and
upregulation of GLS1 expression (180) (Fig.
2), which has been confirmed to serve a key role in the
induction of Snail-dependent EMT and promotion of tumor metastasis
(181). c-Myc can also promote
glycolysis via direct induction of the expression of
glycolytic-associated enzymes, including hexokinase 2 and
phosphoinositide-dependent protein kinase-1 (182), which promotes stemness and EMT
phenotypes in cancer cells (183).
c-Myc-mediated metabolic reprogramming is largely achieved by
effects on mitochondria. The mutual interplay between mitochondrial
metabolism and EMT has been fully addressed (184). Recently, 13C-labeled
metabolite tracer detection revealed that c-Myc-driven lymphoma
cells are highly dependent on mitochondrial aerobic metabolism
(185). In addition, c-Myc can
promote mitochondrial biogenesis and maintain the number and
function of mitochondria in tumor cells (Fig. 2).
IDH
IDH is responsible for catalyzing the metabolism of
isocitrate into α-KG. IDH in mitochondria employs NAD+
as a coenzyme, whereas IDH in the cytoplasm uses NADP+
as a coenzyme, although both catalyze the same reaction. A high
frequency of mutations in IDH1 and IDH2 occurs in glioma (186). Furthermore, IDH mutations are
found in patients with acute myeloid leukemia and myelodysplastic
syndrome (187). A high frequency
of IDH mutation sites associated with tumors are located at the
active site of IDH1 or IDH2, and mutation hotspots include the IDH1
gene R132 locus and IDH2 gene R172 locus. Mutations in both sites
reduce the ability of IDH to catalyze the conversion of isocitrate
to α-KG, whereas the ability to reduce α-KG is enhanced, generating
2-hydroxyglutarate (2HG), which is considered a potential
carcinogenic metabolite (171,188). Because mutations in IDH almost
always occur on one allele, with the other remaining wild type, a
single IDH allele mutation does not seriously affect the ability of
IDH to convert isocitrate to α-KG. The most serious consequences
are caused by the accumulation of 2HG, a by-product of the TCA
cycle, which is present in normal cells at a very low level. The
mechanism by which mutated IDH leads to tumor occurrence has
various explanations. It has been reported that mutation of IDH1 in
astrocytes is sufficient to cause hypermethylation of the genome,
which is commonly found in glioma and other tumors (189). A previous study regarding leukemia
also revealed that IDH mutations can lead to an increase in 2HG
production as a result of TET2 inactivation, causing genome
hypermethylation (190). However,
another study demonstrated that AGH-5198, which specifically
inhibits mutations at the R132H site, can suppress the growth of
glioma cells without causing significant alterations in genomic
methylation levels (191). This
finding suggests that IDH mutation not only leads to genomic
epigenetic alterations but possibly additional mechanisms to
promote tumorigenesis. In addition, IDH1 mutations lead to EMT
through upregulation of the transcription factor ZEB1 and
downregulation of the miR-200 family of miRs (192). Furthermore, the oncometabolite 2HG
can directly induce EMT and distant metastasis in colorectal cancer
(168).
Conclusion and prospects
Metabolic reprogramming is an essential pathway for
genetic events that mediate malignant transformation, including
EMT, thus promoting tumor metastasis. Aberrant expression of genes
including Kras, p53, c-Myc and IDH induces metabolic reprogramming.
Through subversive metabolic adjustment, tumor cells are provided
access to the required energy supply, and balancing the energy
supply and synthesis of biological macromolecules is important to
achieve rapid proliferation of cell populations. Inducing EMT not
only promotes the proliferation of tumor cells from the primary
site, but also enhances the self-renewal ability of tumor cells.
EMT can transform tumor cells into CSCs, and CSCs may acquire
migratory ability through EMT transformation, in order to help
tumor cells escape drug treatment. This review summarized the
interactions between tumor metabolic reprogramming and EMT in tumor
cells, aiming to provide insights into targeting key metabolic
molecules for the treatment of tumor metastasis.
Acknowledgements
Not applicable.
Funding
The present study was financially sponsored by
grants from the National Natural Science Foundation of China (grant
nos. 31401161, 81502370 and 81702845), the State Key Laboratory of
Cancer Biology Project (grant no. CBSKL2017Z11) and the Special
Program of Shaanxi Educational Commission (grant no. 17JK0220).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
ML, XB, BC, XQ and LS performed the literature
search and wrote the paper. PL and KL performed the literature
search. XQ and LS approved the final version of the manuscript to
be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer's Achilles' heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mayevsky A: Mitochondrial function and
energy metabolism in cancer cells: Past overview and future
perspectives. Mitochondrion. 9:165–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Walenta S and Mueller-Klieser WF: Lactate:
Mirror and motor of tumor malignancy. Semin Radiat Oncol.
14:267–274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakamoto S, Tsukada K, Sagara T, Kawachi
Y, Murakami S and Iwama T: Human colorectal malignancy and oral
UFT. Anticancer Res. 22:339–341. 2002.PubMed/NCBI
|
|
5
|
Santos CR and Schulze A: Lipid metabolism
in cancer. FEBS J. 279:2610–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Furuta E, Okuda H, Kobayashi A and Watabe
K: Metabolic genes in cancer: Their roles in tumor progression and
clinical implications. Biochim Biophys Acta. 1805:141–152.
2010.PubMed/NCBI
|
|
7
|
Martin RM, Vatten L, Gunnell D, Romundstad
P and Nilsen TI: Components of the metabolic syndrome and risk of
prostate cancer: The HUNT 2 cohort, Norway. Cancer Causes Control.
20:1181–1192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siegel AB and Zhu AX: Metabolic syndrome
and hepatocellular carcinoma: Two growing epidemics with a
potential link. Cancer. 115:5651–5661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stocks T, Lukanova A, Johansson M, Rinaldi
S, Palmqvist R, Hallmans G, Kaaks R and Stattin P: Components of
the metabolic syndrome and colorectal cancer risk; A prospective
study. Int J Obes. 32:304–314. 2008. View Article : Google Scholar
|
|
10
|
Kabat GC, Kim M, Chlebowski RT, Khandekar
J, Ko MG, McTiernan A, Neuhouser ML, Parker DR, Shikany JM,
Stefanick ML, et al: A longitudinal study of the metabolic syndrome
and risk of postmenopausal breast cancer. Cancer Epidemiol
Biomarkers Prev. 18:2046–2053. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Foroni C, Broggini M, Generali D and Damia
G: Epithelial-mesenchymal transition and breast cancer: Role,
molecular mechanisms and clinical impact. Cancer Treat Rev.
38:689–697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiery JP: Epithelial-mesenchymal
transition in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Christofori G and Semb H: The role of the
cell-adhesion molecule E-cadherin a tumour-suppresor gene. Trends
Biochemci Sci. 24:73–76. 1999. View Article : Google Scholar
|
|
16
|
Hiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boyer B, Valls AM and Edme N: Induction
and regulation of epithelial-mesenchymal transitions. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saitoh M and Miyazawa K: Transcriptional
and post-transcriptional regulation in TGF-β-mediated
epithelial-mesenchymal transition. J Biochem. 151:563–71. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gradl D, Kuhl M and Wedlich D: The Wnt/Wg
signal transducer beta-catenin controls fibronectin expression. Mol
Cell Biol. 19:5576–5587. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gilles C, Polette M, Mestdagt M,
Nawrocki-Raby B, Ruggeri P, Birembaut P and Foidart JM:
Transactivation of vimentin by beta-catenin in human breast cancer
cells. Cancer Res. 63:2658–2664. 2003.PubMed/NCBI
|
|
22
|
Wang Z, Li Y, Kong D and Sarkar FH: The
role of Notch signaling pathway in epithelial-mesenchymal
transition (EMT) during development and tumor aggressiveness. Curr
Drug Targets. 11:745–751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kume T: The role of FoxC2 transcription
factor in tumor angiogenesis. J Oncol. 2012:2045932012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Harris TJ and Tepass U: Adherens
junctions: From molecules to morphogenesis. Nat Rev Mol Cell Biol.
11:502–514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola
D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH: Twist is
transcriptionally induced by activation of STAT3 and mediates STAT3
oncogenic function. J Biol Chem. 283:14665–14673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cairns R, Harris IS and Mak TW: Regulation
of cancer cell metabolism. Nat Rev Cancer. 11:85–95. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinberg R: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kaartinen V, Voncken JW, Shuler C,
Warburton D, Bu D, Heisterkamp N and Groffen J: Abnormal lung
development and cleft palate in mice lacking TGF-beta-3 indicates
defects of epithelial-mesenchymal interaction. Nat Genet.
11:415–421. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khoury H, Dankort DL, Sadekova S, Naujokas
MA, Muller WJ and Park M: Distinct tyrosine autophosphorylation
sites mediate induction of epithelial mesenchymal like transition
by an activated ErbB-2/Neu receptor. Oncogene. 20:788–799. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lester RD, Jo M, Montel V, Takimoto S and
Gonias SL: uPAR induces epithelial-mesenchymal transition in
hypoxic breast cancer cells. J Cell Biol. 178:425–436. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gheldof A and Berx G: Cadherins and
epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci.
116:317–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takai E, Tan X, Tamori Y, Hirota M, Egami
H and Ogawa M: Correlation of translocation of tight junction
protein Zonula occludens-1 and activation of epidermal growth
factor receptor in the regulation of invasion of pancreatic cancer
cells. Int J Oncol. 27:645–651. 2005.PubMed/NCBI
|
|
36
|
Ryeom SW, Paul D and Goodenough DA:
Truncation mutants of the tight junction protein ZO-1 disrupt
corneal epithelial cell morphology. Mol Biol Cell. 11:1687–1697.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Radisky DC, Levy DD, Littlepage LE, Liu H,
Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et
al: Rac1b and reactive oxygen species mediate MMP-3-induced EMT and
genomic instability. Nature. 436:123–127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miyoshi A, Kitajima Y, Sumi K, Sato K,
Hagiwara A, Koga Y and Miyazaki K: Snail and SIP1 increase cancer
invasion by upregulating MMP family in hepatocellular carcinoma
cells. Br J Cancer. 90:1265–1273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang L, Sugiura H, Sugiura H, Huang X,
Ali A, Kuro-o M, Deberardinis RJ and Boothman DA: Metabolic
reprogramming during TGFβ1-induced epithelial-to-mesenchymal
transition. Oncogene. 34:3908–3916. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang H, Liu CY, Zha ZY, Zhao B, Yao J,
Zhao S, Xiong Y, Lei QY and Guan KL: TEAD transcription factors
mediate the function of TAZ in cell growth and
epithelial-mesenchymal transition. J Biol Chem. 284:13355–13362.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sahlgren C, Gustafsson MV, Jin S,
Poellinger L and Lendahl U: Notch signaling mediates
hypoxia-induced tumor cell migration and invasion. Proc Natl Acad
Sci USA. 105:6392–6397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zheng H and Kang Y: Multilayer control of
the EMT master regulators. Oncogene. 33:1755–1763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The role of snail in EMT and tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin CY, Tsai PH, Kandaswami CC, Lee PP,
Huang CJ, Hwang JJ and Lee MT: Matrix metalloproteinase-9
cooperates with transcription factor Snail to induce
epithelial-mesenchymal transition. Cancer Sci. 102:815–827. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Haraguchi M, Sato M and Ozawa M:
CRISPR/Cas9n-mediated deletion of the snail 1gene (SNAI1)
reveals its role in regulating cell morphology, cell-cell
interactions, and gene expression in ovarian cancer (RMG-1) cells.
PLoS One. 10:e01322602015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim NH, Cha YH, Lee J, Lee SH, Yang JH,
Yun JS, Cho ES, Zhang X, Nam M, et al: Snail reprograms glucose
metabolism by repressing phosphofructokinase PFKP allowing cancer
cell survival under metabolic stress. Nat Commun. 8:143742017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang F, Sun L, Li Q, Han X, Lei L, Zhang H
and Shang Y: SET8 promotes epithelial-mesenchymal transition and
confers TWIST dual transcriptional activities. EMBO J. 31:110–123.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang
Z, Yang CJ, Yuan L and Ouyang G: Twist2 contributes to breast
cancer progression by promoting an epithelial-mesenchymal
transition and cancer stem-like cell self-renewal. Oncogene.
30:4707–4720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tania M, Khan MA and Fu J: Epithelial to
mesenchymal transition inducing transcription factors and
metastatic cancer. Tumour Biol. 35:7335–7342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gemmill RM, Roche J, Potiron VA, Nasarre
P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA and
Drabkin HA: ZEB1-responsive genes in non-small cell lung cancer.
Cancer Lett. 300:66–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Halldorsson S, Rohatgi N, Magnusdottir M,
Choudhary KS, Gudjonsson T, Knutsen E, Barkovskaya A, Hilmarsdottir
B, Perander M, Mælandsmo GM, et al: Metabolic re-wiring of isogenic
breast epithelial cell lines following epithelial to mesenchymal
transition. Cancer Lett. 396:117–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Anastasiou D, Poulogiannis G, Asara JM,
Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW,
Auld DS, et al: Inhibition of pyruvate kinase M2 by reactive oxygen
species contributes to cellular antioxidant responses. Science.
334:1278–1283. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dong C, Yuan T, Wu Y, Wang Y, Fan TW,
Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al: Loss of FBP1 by
Snail-mediated repression provides metabolic advantages in
Basal-like breast cancer. Cancer Cell. 23:316–331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Warburg O, Posener K and Negelein E: Ueber
den stoffwechsel der tumoren. Biochemische Zeitschrif. 152:319–344.
1924.(In German).
|
|
57
|
Porporato PE, Payen VL, Perez-Escuredo J,
De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T,
Bouzin C, et al: A mitochondrial switch promotes tumor metastasis.
Cell Rep. 8:754–766. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wood T: Physiological functions of the
pentose phosphate pathway. Cell Biochem Funct. 4:241–247. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Schieber MS and Chandel NS: ROS links
glucose metabolism to breast cancer stem cell and EMT phenotype.
Cancer Cell. 23:265–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu HW, Zhu X, Zhang J, Zhang XB and Tan
W: A red emitting two-photon fluorescent probe for dynamic imaging
of redox balance meditated by a superoxide anion and GSH in living
cells and tissues. Analyst. 141:5893–5899. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie
MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al: Association
of reactive oxygen species levels and radioresistance in cancer
stem cells. Nature. 458:780–783. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Locasale JW: Serine, glycine and
one-carbon units: Cancer metabolism in full circle. Nat Rev Cancer.
13:572–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Luka Z, Mudd SH and Wagner C: Glycine
N-methyltransferase and regulation of
S-adenosylmethionine levels. J Biol Chem. 284:22507–22511.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
McCabe MT, Ott HM, Ganji G, Korenchuk S,
Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III,
Diaz E, et al: EZH2 inhibition as a therapeutic strategy for
lymphoma with EZH2-activating mutations. Nature. 492:108–112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cook CC, Kim A, Terao S, Gotoh A and
Higuchi M: Consumption of oxygen: A mitochondrial generated
progression signal of advanced cancer. Cell Death Dis. 3:e2582012.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chiche J, Rouleau M, Gounon P,
Brahimi-Horn MC, Pouysségur J and Mazure NM: Hypoxic enlarged
mitochondria protect cancer cells from apoptotic stimuli. J Cell
Physiol. 222:648–657. 2010.PubMed/NCBI
|
|
67
|
Kamarajugadda S, Stemboroski L, Cai Q,
Simpson NE, Nayak S, Tan M and Lu J: Glucose oxidation modulates
anoikis and tumor metastasis. Mol Cell Biol. 32:1893–1907. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Andersen JL and Kornbluth S: Mcl-1 rescues
a glitch in the matrix. Nat Cell Biol. 14:563–565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Morandi A, Taddei ML, Chiarugi P and
Giannoni E: Targeting the metabolic reprogramming that controls
epithelial-to-mesenchymal transition in aggressive tumors. Front
Oncol. 7:402017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bonuccelli G, Tsirigos A, Whitaker-Menezes
D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N,
Howell A, Martinez-Outschoorn UE, et al: Ketones and lactate ‘fuel’
tumor growth and metastasis: Evidence that epithelial cancer cells
use oxidative mitochondrial metabolism. Cell Cycle. 9:3506–3514.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Doherty JR and Cleveland JL: Targeting
lactate metabolism for cancer therapeutics. J Clin Invest.
123:3685–3692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu M, Quek LE, Sultani G and Turner N:
Epithelial-mesenchymal transition induction is associated with
augmented glucose uptake and lactate production in pancreatic
ductal adenocarcinoma. Cancer Metab. 4:192016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Goetze K, Walenta S, Ksiazkiewicz M,
Kunz-Schughart LA and Mueller-Klieser W: Lactate enhances motility
of tumor cells and inhibits monocyte migration and cytokine
release. Int J Oncol. 39:453–463. 2011.PubMed/NCBI
|
|
75
|
Sotnik JL, Lori JC, Rose BJ and Thamm DH:
Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic
phenotype in vitro and in vivo. Clin Exp Metastasis. 28:865–875.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
David CJ, Chen M, Assanah M, Canoll P and
Manley JL: HnRNP proteins controlled by c-Myc deregulate pyruvate
kinase mRNA splicing in cancer. Nature. 463:364–368. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Christofk HR, Vander HM, Wu N, Asara JM
and Cantley LC: Pyruvate kinase M2 is a phosphotyrosine-binding
protein. Nature. 452:181–186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hittosugi T, Kang S, Vander Heiden MG,
Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et
al: Tyrosine phosphorylation inhibits PKM2 to promote the Warburg
effect and tumor growth. Sci Signal. 2:ra732009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:232–744. 2011. View Article : Google Scholar
|
|
81
|
Elisa G, Maria LT, Andrea M, Comito G,
Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D and
Chiarugi P: Targeting stromal-induced pyruvate kinase M2 nuclear
translocation impairs oxphos and prostate cancer metastatic spread.
Oncotarget. 6:24061–24074. 2015.PubMed/NCBI
|
|
82
|
Zhan C, Shi Y, Lu C and Wang Q: Pyruvate
kinase M2 is highly correlated with the differentiation and the
prognosis of esophageal squamous cell cancer. Dis Esophagus.
26:746–753. 2013.PubMed/NCBI
|
|
83
|
Li J, Yang Z, Zou Q, Yuan Y, Li J, Liang
L, Zeng G and Chen S: PKM2 and ACVR 1C are prognostic markers for
poor prognosis of gallbladder cancer. Clin Transl Oncol.
16:200–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Feng C, Gao Y, Wang C, Yu X, Zhang W, Guan
H, Shan Z and Teng W: Aberrant overexpression of pyruvate kinase M2
is associated with aggressive tumor features and the BRAF
mutation in papillary thyroid cancer. J Clin Endocrinol Metab.
98:E1524–E1533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Goldberg MS and Sharp PA: Pyruvate kinase
M2-specific siRNA induces apoptosis and tumor regression. J Exp
Med. 209:217–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yoo YG, Christensen J, Gu J and Huang LE:
HIF-1α mediates tumor hypoxia to confer a perpetual mesenchymal
phenotype for malignant progression. Sci Signal. 4:pt42011.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network down
stream of m TOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Icard P, Kafara P, Steyaert JM, Schwartz L
and Lincet H: The metabolic cooperation between cells in solid
cancer tumors. Biochim Biophys Acta. 1846:216–225. 2014.PubMed/NCBI
|
|
89
|
Wu CY, Tsai YP, Wu MZ, Teng SC and Wu KJ:
Epigenetic reprogramming and post-transcriptional regulation during
the epithelial-mesenchymal transition. Trends Genet. 28:454–463.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Song IS, Wang AG, Yoon SY, Kim JM, Kim JH,
Lee DS and Kim NS: Regulation of glucose metabolism-related genes
and VEGF by HIF-1alpha and HIF-1beta, but not HIF-2alpha, in
gastric cancer. Exp Mol Med. 41:51–58. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhao T, Zhu Y, Morinibu A, Kobayashi M,
Shinomiya K, Itasaka S, Yoshimura M, Guo G, Hiraoka M and Harada H:
HIF-1-mediated metabolic reprogramming reduces ROS levels and
facilitates the metastatic colonization of cancers in lungs. Sci
Rep. 4:37932014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Psaila B and Lyden D: The metastatic
niche: Adapting the foreign soil. Nat Rev Cancer. 9:285–293. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Erler JT, Bennewith KL, Cox TR, Lang G,
Bird D, Koong A, Le QT and Giaccia AJ: Hypoxia-induced lysyl
oxidase is a critical mediator of bone marrow cell recruitment to
form the premetastatic niche. Cancer Cell. 15:35–44. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ippolito L, Marini A, Cavallini L, Morandi
A, Pietrovito L, Pintus G, Giannoni E, Schrader T, Puhr M, Chiarugi
P and Taddei ML: Metabolic shift toward oxidative phosphorylation
in docetaxel resistant prostate cancer cells. Oncotarget.
7:61890–61904. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mashek DG and Coleman RA: Cellular fatty
acid uptake: The contribution of metabolism. Curr Opin Lipidol.
17:274–278. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zechner R, Strauss JG, Haemmerle G, Lass A
and Zimmermann R: Lipolysis: Pathway under construction. Curr Opin
Lipidol. 16:333–340. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Menendez JA, Vellon L, Oza BP and Lupu R:
Does endogenous fatty acid metabolism allow cancer cells to sense
hypoxia and mediate hypoxic vasodilatation? Characterization of a
novel molecular connection between fatty acid synthase (FAS) and
hypoxia-inducible factor-1alpha (HIF-1alpha)-related expression of
vascular endothelial growth factor (VEGF) in cancer cells
overexpressing her-2/neu oncogene. J Cell Biochem. 94:857–863.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Swierczyński J and Sledziński T: Metabolic
and regulatory function of fatty acid synthase. Postepy Biochem.
58:175–185. 2012.(In Polish). PubMed/NCBI
|
|
100
|
Jeong NY, Lee JS, Yoo KS, Oh S, Choe E,
Lee HJ, Park BS, Choi YH and Yoo YH: Fatty acid synthase inhibitor
cerulenin inhibits topoisomerase I catalytic activity and augments
SN-38-induced apoptosis. Apoptosis. 18:226–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Fujita Y, Krause G, Scheffner M, Zechner
D, Leddy HE, Behrens J, Sommer T and Birchmeier W: Hakai, a
c-Cbl-like protein, ubiquitinates and induces endocytosis of the
E-cadherin complex. Nat Cell Biol. 4:222–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li J, Dong L, Wei D, Wang X, Zhang S and
Li H: Fatty acid synthase mediates the epithelial-mesenchymal
transition of breast cancer cells. Int J Biol Sci. 10:171–180.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rakheja D, Kapur P, Hoang MP, Roy LC and
Bennett MJ: Increased ratio of saturated to unsaturated C18 fatty
acids in colonic adenocarcinoma: Implications for cryotherapy and
lipid raft function. Med Hypotheses. 65:1120–1123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ikonnen E: Roles of lipid rafts in
membrane transport. Curr Oppin Cell Biol. 13:470–477. 2001.
View Article : Google Scholar
|
|
105
|
Russell DW: The enzymes, regulation, and
genetics of bile acid synthesis. Annu Rev Biochem. 72:137–174.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pelton K, Freeman MR and Solomon KR:
Cholesterol and cancer. Curr Opin Pharmacol. 12:751–759. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kitahara CM, Berrington de González A,
Freeman ND, Huxley R, Mok Y, Jee SH and Samet JM: Total cholesterol
and cancer risk in a large prospective study in Korea. J Clin
Oncol. 29:1592–1598. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Alikhani N, Ferguson RD, Novosyadlyy R,
Gallagher EJ, Scheinman EJ, Yakar S and LeRoith D: Mammary tumor
growth and pulmonary metastasis are enhanced in a hyperlipidemic
mouse model. Oncogene. 32:961–967. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Nielsen SF, Nordestgaard BG and Bojesen
SE: Statin use and reduced cancer-related mortality. N Engl J Med.
367:1792–1802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Taras D, Blanc JF, Rullier A, Dugot-Senant
N, Laurendeau I, Vidaud M and Rosenbaum J: Pravastatin reduces lung
metastasis of rat hepatocellular carcinoma via a coordinated
decrease of MMP expression and activity. J Hepatol. 46:69–76. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhang J, Yang Z, Xie L, Xu L, Xu D and Liu
X: Statins, autophagy and cancer metastasis. Int J Biochem Cell
BIol. 45:745–752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Roy M, Kung HJ and Ghosh PM: Statins and
prostate cancer: Role of cholesterol inhibition vs. prevention of
small GTP-binding proteins. Am J Cancer Res. 1:542–561.
2011.PubMed/NCBI
|
|
113
|
Simons K and Ikonen E: Functional rafts in
cell membranes. Nature. 387:569–572. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Freeman MR, Di Vizio D and Solomon KR: The
Rafts of the Medusa: Cholesterol targeting in cancer therapy.
Oncogene. 29:3745–3747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Patra SK: Dissecting lipid raft
facilitated cell signaling pathways incancer. Biochim Biophys Acta.
1785:182–206. 2008.PubMed/NCBI
|
|
116
|
Mural T: The role of lipid rafts in cancer
cell adhesion and migration. Int J Cell Biol.
2012:7632832012.PubMed/NCBI
|
|
117
|
Rangaswami H, Bulbule A and Kundu GC:
Osteopontin: Role in cell signaling and cancer progression. Trends
Cell Biol. 16:79–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Murai T, Maruyama Y, Mio K, Nishiyama H,
Suga M and Sato C: Low cholesterol triggers membrane
microdomain-dependent CD44 shedding and suppresses tumor cell
migration. J Biol Chem. 286:1999–2007. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37 Suppl 4:S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Irwin ME, Mueller KL, Bohin N, Ge Y and
Boerner JL: Lipid raft localization of EGFR alters the response of
cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J
Cell Physiol. 226:2316–2328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Afshordel S, Kern B, Clasohm J, König H,
Priester M, Weissenberger J, Kögel D and Eckert GP: Lovastatin and
perillyl alcohol inhibit glioma cell invasion, migration, and
proliferation - impact of Ras-/Rho-prenylation. Pharmacol Res.
91:69–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Nelson ER, Wardell SE, Jasper JS, Park S,
Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V,
et al: 27-Hydroxycholesterol links hypercholesterolemia and breast
cancer pathophysiology. Science. 342:1094–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wu Q, Ishikawa T, Sirianni R, Tang H,
McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA,
et al: 27-Hydroxycholesterol promotes cell-autonomous, ER-positive
breast cancer growth. Cell Rep. 5:637–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Hensley CT, Wasti AT and Deberardinis RJ:
Glutamine and cancer: Cell biology, physiology and clinical
opportunities. J Clin Invest. 123:3678–3684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kim D, Fiske BP, Birsoy K, Freinkman E,
Kami K, Possemato RL, Chudnovsky Y, Pacold ME, Chen WW, Cantor JR,
et al: SHMT2 drives glioma cell survival in ischaemia but imposes a
dependence on glycine clearance. Nature. 520:363–367. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Roberts E and Frankel S: Free amino acid
in normal and neoplastic tissues of mice as studied by paper
chromatography. Cancer Res. 9645–648. (3 pl)1949.PubMed/NCBI
|
|
127
|
Roberts E and Borges PR: Patterns of free
amino acids in growing and regressing tumor. Cancer Res.
15:697–699. 1955.PubMed/NCBI
|
|
128
|
Yuneva M, Zamboni N, Oefner P,
Sachidanandam R and Lazebnik Y: Deficiency in glutamine but not
glucose induces MYC-dependent apoptosis in human cells. J Cell
Biol. 178:93–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wise DR, Deberardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB, et al: Myc regulates a transcriptional program that stimulates
mitochondrial glutaminolysis and leads to glutamine addiction. Proc
Natl Acad Sci USA. 105:18782–18787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wang JB, Erickson JW, Fuji R, Ramachandran
S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et
al: Targeting mitochondrial glutaminase activity inhibits oncogenic
transformation. Cancer Cell. 18:207–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Jin L, LI D, Alesi GN, Fan J, Kang HB, Lu
Z, Boggon TJ, Jin P, Yi H, Wright ER, et al: Glutamate
dehydrogenase1 signals through antioxidant glutathione peroxidase1
to regulate redox homeostasis and tumor growth. Cancer Cell.
27:257–270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Piskounova E, Agathocleous M, Murphy MM,
Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ
and Morrison SJ: Oxidative stress inhibits distant metastasis by
human melanoma cells. Nature. 527:186–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Kung HN, Marks JR and Chi JT: Glutamine
synthetase is a genetic determinant of cell type-specific glutamine
independence in breast epithelia. PLoS Genet. 7:e10022292011.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Maher EA, Marin-Valencia I, Bachoo RM,
Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C,
Madden C, et al: Metabolism of [U-13 C]glucose in human
brain tumors in vivo. NMR Biomed. 25:1234–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Guohua C, Stephanie L and Jian W: Glycine
decarboxylase is a target for transcriptional repressor Snail.
Cancer Metab. 2 Suppl 1:P832014. View Article : Google Scholar :
|
|
137
|
Lepage GA: In vitro incorporation of
glycine-2-C14 into purines and proteins. Cancer Res.
13:178–185. 1953.PubMed/NCBI
|
|
138
|
Zhang WC, Shyh-Chang N, Yang H, Rai A,
Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, et al: Glycine
decarboxylase activity drives non-small cell lung cancer
tumor-initiating cells and tumorigenesis. Cell. 148:259–272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Shen T: Biochemistry. 2nd. Beijing: Higher
Education Press; pp. 270–273. 1991, PubMed/NCBI
|
|
140
|
Rose ML, Madren J, Bunzendahl H and
Thurman RG: Dietary glycine inhibits the growth of B16 melanoma
tumors in mice. Carcinogenesis. 20:793–798. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kalhan SC and Hanson RW: Resurgence of
serine: An often neglected but indispensable amino acid. J Biol
Chem. 287:19786–19791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Possemato R, Marks KM, Shaul YD, Pacold
ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et
al: Functional genomics reveal that the serine synthesis pathway is
essential in breast cancer. Nature. 476:346–350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Pipkorn R, Wiessler M, Waldeck W, Hennrich
U, Nokihara K, Beining M and Braun K: Improved synthesis strategy
for peptide nucleic acids (PNA) appropriate for cell-specific
fluorescence imaging. Int J Med Sci. 9:1–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lin J, Teo S, Lam DH, Jeyaseelan K and
Wang S: MicroRNA-10b pleiotropically regulates invasion,
angiogenicity and apoptosis of tumor cells resembling mesenchymal
subtype of glioblastoma multiforme. Cell Death Dis. 3:e3982012.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhang C, Zhang Z, Zhu Y and Qin S:
Glucose-6-phosphate dehydrogenase: A biomarker and potential
therapeutic target for cancer. Anticancer Agents Med Chem.
14:280–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wang R, Yu C, Zhao D, Wu M and Yang Z: The
mucin-type glycosylating enzyme polypeptide
N-acetylgalactosaminyltransferase 14 promotes the migration of
ovarian cancer by modifying mucin 13. Oncol Rep. 30:667–676. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Li Y, Liu H, Huang YY, Pu LJ, Zhang XD,
Jiang CC and Jiang ZW: Suppression of endoplasmic reticulum
stress-induced invasion and migration of breast cancer cells
through the downregulation of heparanase. Int J Mol Med.
31:1234–1242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lin RZ, Wang TP, Hung RJ, Chuang YJ, Chien
CC and Chang HY: Tumor-induced endothelial cell apoptosis: Roles of
NAD(P)H oxidase-derived reactive oxygen species. J Cell Physiol.
226:1750–1762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Qian YR, Guo Y, Wan HY, Fan L, Feng Y, Ni
L, Xiang Y and Li QY: Angiotensin-converting enzyme 2 attenuates
the metastasis of non-small cell lung cancer through inhibition of
epithelial mesenchymal transition. Oncol Rep. 29:2408–2414. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kim EK, Park JM, Lim S, Choi JW, Kim HS,
Seok H, Seo JK, Oh K, Lee DS, Kim KT, et al: Activation of
AMP-activated protein kinase is essential for lysophosphatidic
acid-induced cell migration in ovarian cancer cells. J Biol Chem.
286:24036–24045. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Qiu SL, Xiao ZC, Piao CM, Xian YL, Jia LX,
Qi YF, Han JH, Zhang YY and Du J: AMP-activated protein kinase α2
protects against liver injury from metastasized tumors via reduced
glucose deprivation-induced oxidative stress. J Biol Chem.
289:9449–9459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Skubitz AP, Taras EP, Boylan KL, Waldron
NN, Oh S, Panoskaltsis-Mortari A and Vallera DA: Targeting CD133 in
an in vivo ovarian cancer model reduces ovarian cancer progression.
Gynecol Oncol. 130:579–587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Chen J, Liu Q, Xiao J and Du J:
EpCAM-antibody-labeled noncytotoxic polymer vesicles for cancer
stem cells-targeted delivery of anticancer drug and siRNA.
Biomacromolecules. 16:1695–1705. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Lawson DA, Bhakta NR, Kessenbrock K,
Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY,
et al: Single-cell analysis reveals a stem-cell program in human
metastatic breast cancer cells. Nature. 526:131–135. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Yang Y, Fan Y, Qi Y, Liu D, Wu K, Wen F
and Zhao S: Side population cells separated from A549 Lung cancer
cell line possess cancer stem cell-like properties and inhibition
of autophagy potentiates the cytotoxic. Oncol Rep. 34:929–935.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Iacopino F, Angelucci C, Piacentini R,
Biamonte F, Mangiola A, Maira G, Grassi C and Sica G: Isolation of
cancer stem cells from three human glioblastoma cell lines:
Characterization of two selected clones. PLoS One. 9:e1051662014.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Aguilar E, Marin de Mas I, Zodda E, Marin
S, Morrish F, Selivanov V, Meca-Cortés Ó, Delowar H, Pons M,
Izquierdo I, et al: Metabolic reprogramming and dependencies
associated with epithelial cancer stem cells independent of the
epithelial-mesenchymal transition program. Stem Cells.
34:1163–1176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Visvader JE and Lindeman GJ: Cancer stem
cells in solid tumors: Accumulating evidence and unresolved
questions. Nat Rev Cancer. 8:755–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Oshimori N, Oristian D and Fuchs E: TGF-β
promotes heterogeneity and drug resistance in squamous cell
carcinoma. Cell. 160:963–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Gammon L, Biddle A, Heywood HK,
Johannessen AC and Mackenzie IC: Sub-sets of cancer stem cells
differ intrinsically in their patterns of oxygen metabolism. PLoS
One. 8:e624932013. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Biddle A, Liang X, Gammon L, Fazil B,
Harper LJ, Emich H, Costea DE and Mackenzie IC: Cancer stem cells
in squamous cell carcinoma switch between two distinct phenotypes
that are preferentially migratory or proliferative. Cancer Res.
71:5317–5326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Biddle A, Gammon L, Liang X, Costea DE and
Mackenzie IC: Phenotypic plasticity determines cancer stem cell
therapeutic resistance in oral squamous cell carcinoma.
EBioMedicine. 4:138–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Shi L, Jackstadt R, Siemens H, Li H,
Kirchner T and Hermeking H: p53-induced miR-15a/16-1 and AP4 form a
double-negative feedback loop to regulate epithelial-mesenchymal
transition and metastasis in colorectal cancer. Cancer Res.
74:532–542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Khew-Goodall Y and Goodall GJ:
Myc-modulated miR-9 makes more metastases. Nat Cell Biol.
12:209–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Colvin H, Nishida N, Konno M, Haraguchi N,
Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K,
et al: Oncometabolite D-2-hydroxyglurate directly induces
epithelial-mesenchymal transition and is associated with distant
metastasis in colorectal cancer. Sci Rep. 8:362892016. View Article : Google Scholar
|
|
169
|
Kondoh H, Lleonart ME, Gil J, Wang J,
Degan P, Peters G, Martinez D, Carnero A and Beach D: Glycolytic
enzymes can modulate cellular life span. Cancer Res. 65:177–185.
2005.PubMed/NCBI
|
|
170
|
Hu J, Liu Z and Wang X: Does TP53 mutation
promote ovarian cancer metastasis to omentum by regulating lipid
metabolism? Med Hypotheses. 81:515–520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Paschka P, Schlenk RF, Gaidzik VI, Habdank
M, Krönke J, Bullinger L, Späth D, Kayser S, Zucknick M, Götze K,
et al: IDH1IDH2 mutations are frequent genetic alterations
in acute myeloid leukemia and confer adverse prognosis in
cytogenetically normal acute myeloid leukemia with NPM1
mutation without FLT3 internal tandem duplication. J Clin
Oncol. 28:3636–3643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Gaglio D, Metallo CM, Gameiro PA, Hiller
K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G and
Chiaradonna F: Oncogenic K-Ras decouples glucose and glutamine
metabolism to support cancer cell growth. Mol Syst Biol. 7:5232011.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Guo JY, Chen HY, Mathew R, Fan J,
Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM,
Karantza V, et al: Activated Ras requires autophagy to maintain
oxidative metabolism and tumorigenesis. Genes Dev. 25:460–470.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Schwartzenberg-Bar-Yoseph F, Armoni M and
Karnieli E: The tumor suppressor p53 down-regulates glucose
transporters GLUT1GLUT4 gene expression. Cancer Res.
64:2627–2633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Bensaad K, Tsuruta A, Selak MA, Vidal MN,
Nakano K, Bartrons R, Gottlieb E and Vousden KH: TIGAR, a
p53-inducible regulator of glycolysis and apoptosis. Cell.
126:107–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Matoba S, Kang JG, Patino WD, Wragg A,
Boehm M, Gavrilova O, Hurley PJ, Bunz F and Hwang PM: p53 regulates
mitochondrial respiration. Science. 312:1650–1653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Li F, Wang Y, Zeller KI, Potter JJ, Wonsey
DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA and Dang CV: Myc
stimulates nuclearly encoded mitochondrial genes and mitochondrial
biogenesis. Mol Cell Biol. 25:6225–6234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Smith AP, Verrecchia A, Fagà G, Doni M,
Perna D, Martinato F, Guccione E and Amati B: A positive role for
Myc in TGFbeta-induced Snail transcription and
epithelial-to-mesenchymal transition. Oncogene. 28:422–430. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH,
Park HG, Han SI and Kang HS: Dlx-2 and glutaminase upregulate
epithelial-mesenchymal transition and glycolytic switch.
Oncotarget. 7:7925–7939. 2016.PubMed/NCBI
|
|
181
|
Liu W, Le A, Hancock C, Lane AN, Dang CV,
Fan TW and Phang JM: Reprogramming of proline and glutamine
metabolism contributes to the proliferative and metabolic responses
regulated by oncogenic transcription factor c-Myc. Proc Natl Acad
Sci USA. 109:8983–8988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Kim JW, Gao P, Liu YC, Semenza GL and Dang
CV: Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively
induce vascular endothelial growth factor and metabolic switches
hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol.
27:7381–7393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen
Q, Wang C and Yin T: Up-regulation of glycolysis promotes the
stemness and EMT phenotypes in gemcitabine-resistant pancreatic
cancer cells. J Cell Mol Med. 21:2055–2067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Guerra F, Guaragnella N, Arbini AA, Bucci
C, Giannattasio S and Moro L: Mitochondrial dysfunction: A novel
potential driver of epithelial-to-mesenchymal transition in cancer.
Front Oncol. 7:2952017. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Murphy TA, Dang CV and Young JD:
Isotopically nonstationary 13C flux analysis of
Myc-induced metabolic reprogramming in B-cells. Metab Eng.
15:206–217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Ward PS, Patel J, Wise DR, Abdel-Wahab O,
Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et
al: The common feature of leukemia-associated IDH1 and IDH2
mutations is a neomorphic enzyme activity converting
alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17:225–234.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Turcan S, Rohle D, Goenka A, Walsh LA,
Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al: IDH1
mutation is sufficient to establish the glioma hypermethylator
phenotype. Nature. 483:479–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS,
Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et
al: Leukemic IDH1 and IDH2 mutations result in a hypermethylation
phenotype, disrupt TET2 function, and impair hematopoietic
differentiation. Cancer Cell. 18:553–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Rohle D, Popovici-Muller J, Palaskas N,
Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B,
Komisopoulou E, et al: An inhibitor of mutant IDH1 delays growth
and promotes differentiation of glioma cells. Science. 340:626–630.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Grassian AR, Lin F, Barrett R, Liu Y,
Jiang W, Korpal M, Astley H, Gitterman D, Henley T, Howes R, et al:
Isocitrate dehydrogenase (IDH) mutations promote a reversible
ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition
(EMT). J Biol Chem. 287:42180–42194. 2012. View Article : Google Scholar : PubMed/NCBI
|














