Introduction
Amyotrophic lateral sclerosis (ALS) is a
neurodegenerative disorder, typically manifesting in adulthood,
marked by the progressive loss of both upper and lower motor
neurons. Despite ongoing gene therapy trials, effective
pharmacological treatments for ALS remain elusive, and the mean
survival is 2-5 years post-diagnosis (1,2). The
genetic landscape of ALS is complex, with a vast array of
sporadically occurring mutations with low penetrance, which poses
challenges for the development of effective treatments (3,4).
Smith et al (5) discovered causative mutations in
annexin A11 (ANXA11), a gene involved in calcium and phospholipid
binding. Subsequent studies across diverse ethnic groups have
confirmed the presence of causative ANXA11 mutations, solidifying
the significance of ANXA11 in ALS pathology (6-16).
The array of neurodegenerative conditions linked to ANXA11
mutations extends beyond ALS, encompassing frontotemporal dementia,
multisystem proteinopathy, Paget's disease, muscular dystrophy,
aphasia and oculopharyngeal muscular dystrophy (7,13,17-19).
The role of ANXA11 in neuronal biology has expanded
since discovery of the association between ANXA11 mutations and
familial and sporadic ALS (5).
ANXA11 is associated with specific ALS-implicated pathways, such as
disrupted liquid-liquid phase separation (LLPS) (20) and RNA and stress granule transport
in the context of calcium biology (10,21,22).
However, the exact role of ANXA11 and its relationship with other
key ALS-linked proteins is yet to be fully understood.
Given that ANX serves critical roles in
phospholipid-binding, cellular trafficking and autophagy and the
presence of a highly conserved orthologous gene (ANXB11) in fruit
flies (Drosophila melanogaster), the present study developed
a novel ALS model that overexpresses human ANXA11 mutations in
Drosophila, a widely used model organism in ALS research
(23-26).
The present study aimed to assess the neurological phenotypes
observed in this model and examine the interactions between ANXA11
and other ALS-associated genes.
Materials and methods
Drosophila stocks
A total of 2,535 were used in experiments. The
following stocks were used elav-Gal4, OK371-Gal4,
UAS-eGFP, ubi-Gal80ts as previously
described (27). Interfering RNA
(IR) stocks UAS-anxB11IR HMS01775, was obtained
from Bloomington Drosophila Stock Centre (BDSC) and GD36186 was
obtained from Vienna Drosophila Resource Center (VDRC)).
UAS-TAR DNA-binding protein wild-type
(TDP43WT (human TDP43 Wild-Type) and
UAS-SOD1G85R (Human SOD1 Gly85Arg mutant) were a
gift from Dr Jemeen Sreedharan (King's College London).
UAS-TBPH was a gift from Dr Frank Hirth (King's College
London).
Human ANXA11 transgenes were generated by PCR
cloning the human open reading frames from previously described
vectors (5) adding a Myc tag at the
C terminal. PCR products were purified with QIAquick PCR
purification kit (Qiagen GmbH), cut with XbaI and
XhoI enzymes and cloned in the corresponding sites in the
multi cloning site in pUAST-attB vectors (a gift from Dr Joe
Bateman (King's College London)]. Presence of the correct ANXA11
mutation was validated by Sanger sequencing performed by Eurofins
Genomics. Insertions were generated by injection in the attP40
Drosophila stocks at the Cambridge Fly Facility (Cambridge, UK)
injection service. The following Oligos from Eurofins Genomics were
used for PCR and cloning (restriction sites are underlined, Myc tag
encoding sequence in lower case, ANXA11 sequences in
italics): Forward, 5'-GACTCGAGATGAGCTACCCTGGCTATCC-3' and
reverse, 5'-AGTCTAGATTAc agatc ctct
tctgagatgagtttttgttcGTCATTGCCACCACAGATCTTCAGC-3'.
Drosophila maintenance and
husbandry
All fly stocks were routinely maintained at 18˚C in
an incubator with 60% humidity and standard 12/12-h light cycle on
a standard fly food mixture of yeast, agar and cornmeal with
nipagin and propionic acid.
Lifespan analysis
Flies were monitored over the course of their life
cycle to quantify death across genotypes as previously described
(27). Briefly, newly eclosed flies
were collected daily for 3 days and transferred to a 29˚C incubator
in batches of 20 flies/vial (equal mix of males and females). Three
times/week the flies were counted and transferred into a new vial
with fresh fly food using CO2 to anaesthetise the flies.
The number of flies still alive was recorded each time and flies
that escaped or were stuck in the food were censored (attributed a
value of 0 on the Day). A dead fly was attributed the value 1 on
the day of death.
Negative geotaxis assay
Flies were kept at 29˚C and age-matched female flies
of the genotypes were placed into empty 70 mm tubes (10
flies/tube). When flies are tapped to the bottom of a vial, they
immediately climb to the top of the vial due to their innate
negative geotaxis abilities (27).
To assess negative geotaxis, flies were tapped to the bottom of the
vial after acclimatization and distance climbed by the flies in
2-min intervals over five trials was measured as previously
described (27). The number of
flies climbing to each cm increment was scored and a genotype mean
was calculated for each time point across five trials. Flies that
jumped or did not perform a vertical climb in one movement burst
were excluded from that trial.
Immunohistochemistry
Whole mount larval ventral nerve cords were
dissected from wandering third instar larva fixed in 4%
paraformaldehyde (Sigma) for 45 min on ice, blocked for 1 h at room
temperature (RT) in Phosphate Buffer Saline (PBS) complemented with
0.2% Tritox-X100 (Sigma) and 10% Normal Goat Serum (NGS, Gibco) and
stained with a rabbit anti-GFP (1:300, cat. no. A11122; Thermo
Fisher Scientific, Inc.) and a mouse monoclonal antibody against
Myc tag (1:100, cat. no. 9E10; Roche Diagnostics, Ltd.).
Additionally, flies were aged for 12 days at 29˚C, sacrificed and
whole brains were dissected, fixed and blocked as aforementioned.
The mouse primary antibody against TDP43 (used at 1:500) was a gift
by Dr Jemeen Sreedharan (King's College London). Brains were imaged
on a Nikon A1R inverted confocal microscope and analysis was
performed in NIS Elements (Nikon 5.21). Secondary antibodies used
were Alexa 555 anti-mouse and Alexa 488 anti-rabbit (Thermo fisher
Scientific, Inc.; cat. nos. A21422 and A11008 and) at 1:200. All
antibodies were diluted in Blocking solution. Primary antibodies
were incubated over night at 4˚C, while secondary antibodies were
incubated 1 h at RT. For TDP43 localization, data was normalised
for cell area and expressed as a ratio of cytoplasmic TDP43 over
nuclear TDP43.
Statistical analysis
All data are presented as mean ± SEM and were
analysed using Microsoft Excel (office 365, www.microsopht.com) and GraphPad Prism 9 (www.graphpad.com). Lifespan was analysed with the
Kaplan-Meyer log-rank (Mantel-Cox) test. One-way ANOVA with
Dunnett's multiple comparisons post hoc test was used for
comparison of ≥3 groups of normally distributed data.
Kruskal-Wallis non-parametric analysis with Dunn's multiple
comparisons test was used for comparison of ≥3 groups of
non-normally distributed data. Two-way ANOVA with Dunnett's
multiple comparisons post hoc test was used for negative geotaxis
assays where ≥2 groups of normally distributed data were confounded
by a third parameter (timepoints). P<0.05 was considered to
indicate a statistically significant difference. All experiments
have been performed at least three times.
Results
Drosophila ANXB11 is structurally
similar to human ANXA11
Sequence comparison revealed that Drosophila
ANXB11 was closely related to human ANXA11 than other
Drosophila genes were, with two splicing isoforms expressing
an ANX protein with a long N-terminal tail (Fig. 1A). While the N-terminus containing
the human mutation hotspot (22)
around aa36-40 is not conserved, other key residues mutated in ALS,
such as G175 and R235, are also conserved in Drosophila
(Fig. 1A).
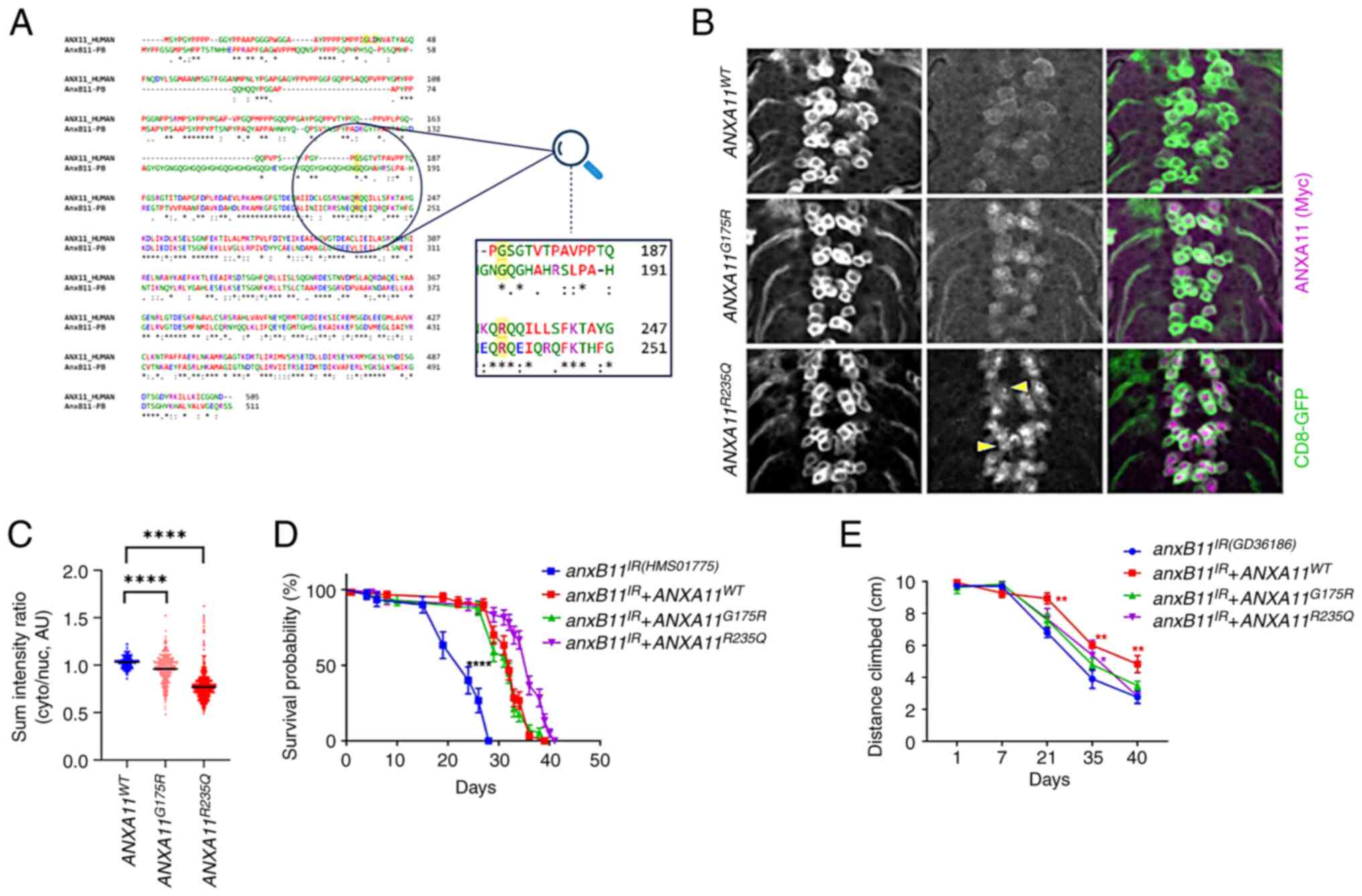 | Figure 1Comparison of human and
Drosophila ANX11. (A) Drosophila ANXB11 is similar to
human ANXA11. G175 and R235 residues are conserved, while the
G38-D40 region is divergent. Numbers indicate the amino acids in
the protein sequence. *Indicates identical aminoacids in the
protein sequence; ***Indicates three identical aminoacids. (B)
Mutant ANXA11 transgenes display nuclear expression in larval motor
neurons. ANXA11R235Q forms discernable aggregates
(arrows). Scale bar, 10 mm. Magnification, x600. (C) Quantification
of nuclear enrichment. ANX11 transgenes rescue the
phenotypes generated by endogenous ANXB11 knockdown for (D)
lifespan and (E) climbing distance. Kaplan-Meier (Log Rank) test.
*P<0.05, **P<0.01,
****P<0.0001 vs. anxB11IR. ANX,
Annexin; IR, Interfering RNAi; WT, Wyld-Type; AU, Arbitrary Units;
cyto, cytoplasm; nuc, nuclear. |
The present study constructed overexpression
transgenes for human ANXA11 with G175R and R235Q mutations, and a
corresponding wild-type (WT) transgene. All transgenes were knocked
into a well-characterised neutral genomic locus to guarantee
similar expression levels and neutrality for insertional
mutagenesis.
ANXA11 transgenes are expressed and
mutants are enriched in the nucleus
The present study demonstrated the expression of
ANXA11 transgenes and notable enrichment of G175R and R235Q mutants
around and within the cell nucleus in larval neurons (Fig. 1B and C). In addition, the R235Q mutant protein
displayed notable aggregation (Fig.
1B), as reported in human cells (5).
ANXA11 transgenes rescue phenotypes
generated by endogenous ANXB11 knockdown
Our previous study showed that knockdown of the fly
ANXB11 gene decreases in lifespan and negative geotaxis (28). The introduction of human ANXA11
transgenes successfully rescued the short lifespan observed
following pan-neuronal knock-down of the endogenous ANXB11
(Fig. 1D). When analysing the
negative geotaxis due to motor neuron knockdown, however, the
rescue was more limited, and only the WT A11 construct constantly
improved negative geotaxis after 21 days (Fig. 1E).
Annexin A11 transgenes improve
lifespan and negative geotaxis defects caused by the expression of
TDP43, but not of Drosophila TBPH
Despite successful expression, altered subcellular
localization and functional alterations of the ANXA11 transgenes,
there were no obvious signs of organism-level toxicity when mutant
ANXA11 transgenes were expressed in Drosophila neurons, both
in terms of lifespan and negative geotaxis. Genetic interactions
with other known ALS genes were assessed using transgenic flies
expressing the human genes that display toxicity in
Drosophila, such as TDP43(24) and SOD1(25).
ANXA11 transgenes markedly worsened lifespan and
negative geotaxis deficits in Drosophila expressing TDP43, a
protein associated with ALS pathology (Fig. 2A and B). ANXA11 transgenes significantly
enhanced the negative geotaxis defects cause by TDP43 after 11 days
and made lifespan significantly shorter.
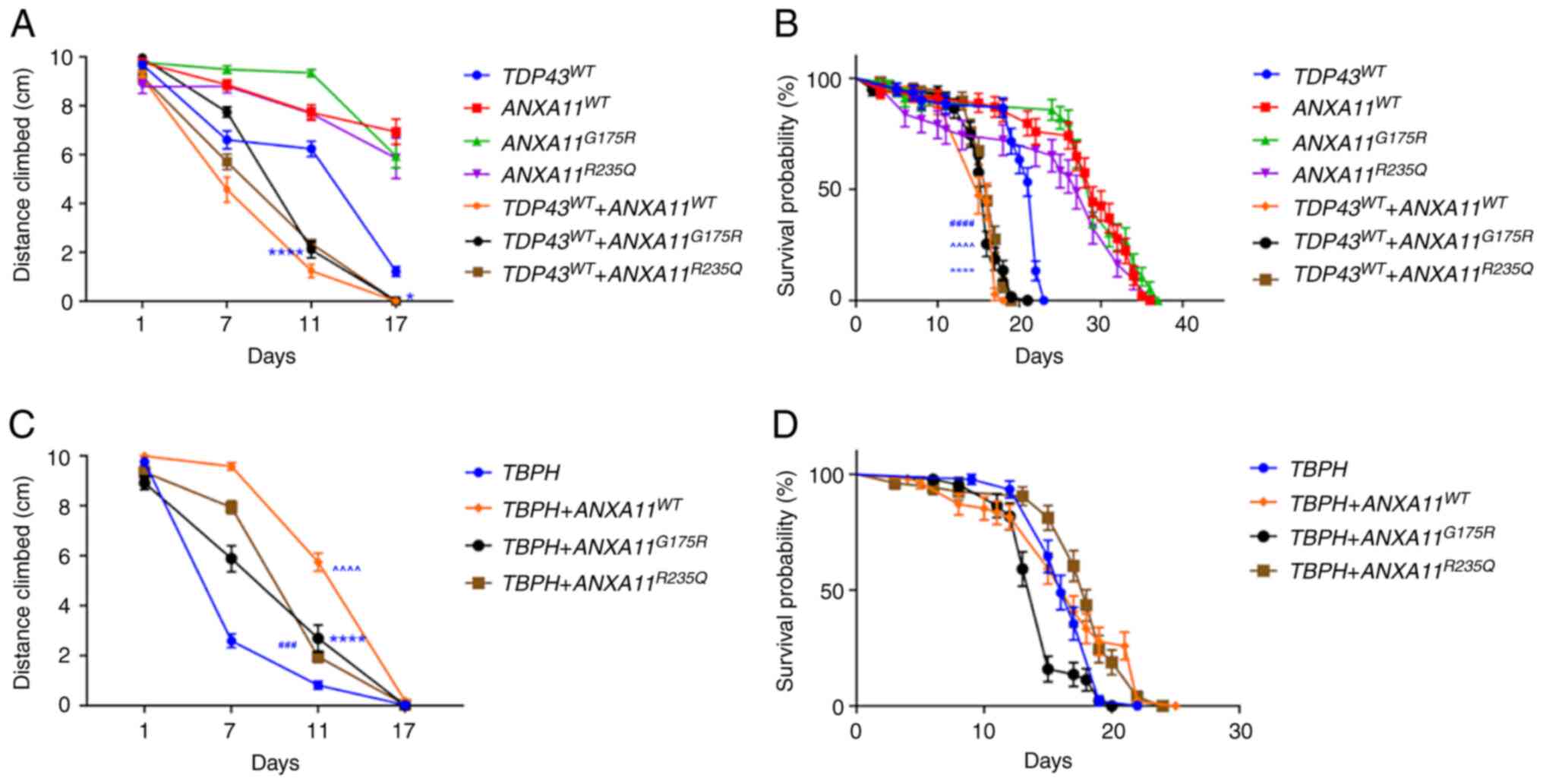 | Figure 2ANXA11 transgenes enhance lifespan
and negative geotaxis defects caused by the expression of TDP43,
but not of Drosophila TBPH. (A) TDP43-expressing flies
illustrate decreased negative geotaxis compared with the ANX
mutants alone, which do not significantly affect negative geotaxis.
When co-expressed with human TDP43, all ANXA11 transgenes
exacerbated the negative geotaxis defects induced by TDP43 alone.
*P= 0.0487, ****P<0.0001 vs. TDP43 WT. (B)
ANXA11 transgenes worsen the lifespan shortening induced by TDP43.
****P<0.0001 vs. TDP43 + ANXA11 WT,
(####P<0.0001 vs. G175R, (^^^^P<0.0001
vs. R235Q)). (C) TBPH induces stronger negative geotaxis defects
alone than when co-expressed with ANX at Day 11.
###P=0.0006 vs. TBPH + ANXA11 G175R and
****P<0.0001 vs. TBPH + ANXA11 R335Q or
(^^^^P<0.0001 vs. ANXA11 WT. (D) ANX did not affect
TBPH lifespan. ANX, Annexin; IR, Interfering RNAi; WT, Wild-Type;
TDP43, TAR DNA binding Protein 43; TBPH, TAR DNA-binding protein-43
homolog. |
Negative geotaxis response and lifespan analysis
showed that OK371-Gal4, TDP43 WT flies illustrate decreased
negative geotaxis compared with the ANX mutants alone, which did
not significantly affect negative geotaxis. When co-expressed with
human TDP43, all ANXA11 transgenes exacerbated the negative
geotaxis defects seen with TDP43 alone (Fig. 2B). Annexin A11 transgenes further
decreased lifespan compared with TDP43 (Fig. 2B). These effects were not observed
in models expressing Drosophila TBPH, an ortholog of TDP43
essential for Drosophila motor neurons (26). TBPH had stronger negative geotaxis
defects alone than when co-expressed with human ANXA11 (Fig. 2C) and ANXA11 did not affect TBPH
lifespan (Fig. 2D).
ANXA11 has no genetic interaction with
human SOD1 in Drosophila
To determine if the modulating effect of ANXA11
transgenes occurred in other ALS-associated genes, the association
between ANXA11 and SOD1 (a gene implicated in familial forms of
ALS) (25) was assessed. There was
no discernible genetic interplay between A11 and G85R mutant SOD1
(Fig. S1).
Localization of human TDP43 is
affected by ANXA11 overexpression and its nuclear increase is
modulated by the mutations in ANXA11
To determine the potential molecular mechanism
underlying the interaction with human TDP43 of the ANXA11
transgenes, the present study assessed expression of TDP43 in the
adult fly brain. Overexpression of ANXA11 altered the typical
distribution of TDP43, increasing its nuclear accumulation
(Fig. 3A and B), which may interfere with key nuclear
functions.
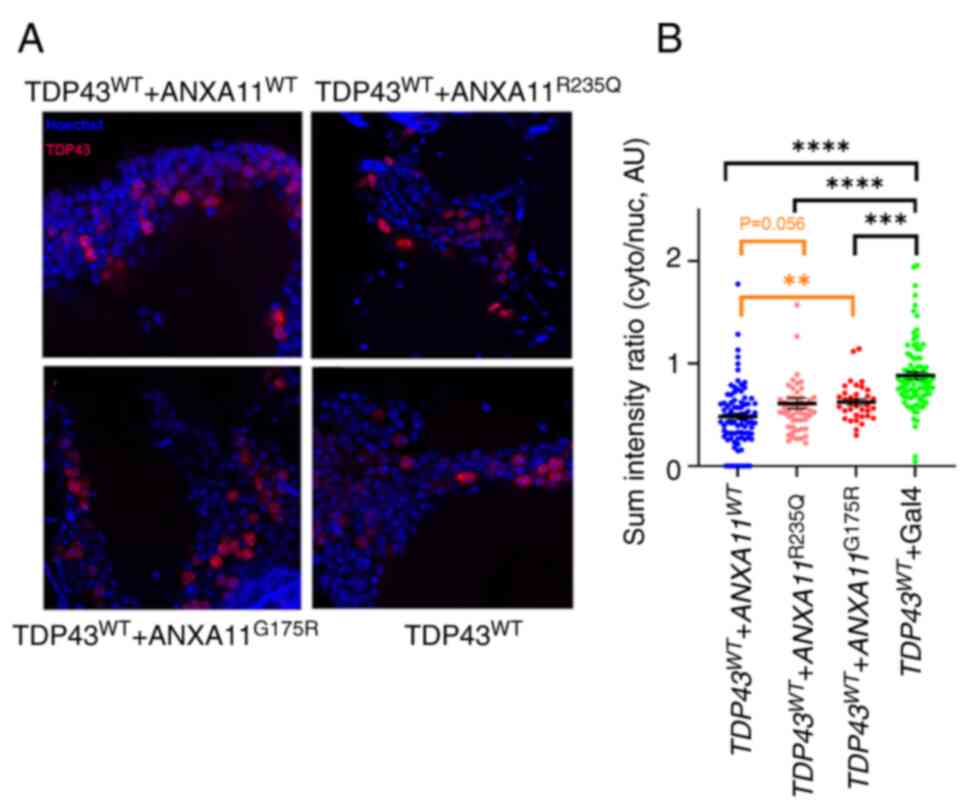 | Figure 3Localization of human TDP43 is
affected by ANX11 overexpression and its nuclear increase is
modulated by the mutations in ANXA11. (A) Expression of
human TDP43 (red) in the brain cells of the fly, showing nuclear
(blue) localisation. Scale bar, 10 mm. Magnification, x600. (B) In
the presence of ANXA11 there is an increased presence of TDP43 in
the nucleus **P<0.01, ***P=0.0002,
****P<0.0001. ANX, annexin; WT, Wild-Type; AU,
arbitrary Units; cyto, cytoplasm; nuc, nuclear; TDP43, TAR DNA
binding Protein 43. |
Furthermore, the extent of TDP43 nuclear
accumulation was decreased by G175R and R235Q mutations in ANXA11
compared with WT ANXA11, indicating that ALS-mutations in ANXA11
may cause promote cytoplasmic localisation of TDP43 compared with
WT ANXA11 (Fig. 3B). These varying
degrees of TDP43 nucleo-cytoplasmic localization suggested that the
link between ANXA11 and TDP43 may be sensitive to the structural
changes induced by these mutations either by gain or loss of
function.
Discussion
The precise mechanism by which ANXA11 causes ALS is
unknown, but progress has been made in understanding the role of
ANXA11 as a molecular tether for axonal RNA transport, which is
impaired by ALS-associated ANXA11 mutations (21). ANXA11 mutations are reported in
patients with sporadic ALS, causing dysregulation of intracellular
Ca2+ homeostasis and stress granule dynamics (10,21).
Drosophila has been widely used as a model
organism for ALS research (23-26)
due to high conservation in ALS genes and ease of genetic
manipulation that allows study of interactions between different
genes. The present study constructed a novel Drosophila
model for studying the role of ANXA11 in ALS pathogenesis.
The present study demonstrated functional
conservation between fly ANXB11 and human ANXA11 and altered
subcellular localisation of ANXA11 mutants. The phenotypical rescue
suggests that the human ANXA11 transgenes not only are expressed
but retain sufficient structural and functional similarity to the
Drosophila ANXB11, allowing them to compensate for its loss.
The more consistent rescue by WT ANXA11 also suggests that the
G175R and the R235Q mutations interfered, at least partially, with
full ANXA11 functionality in Drosophila.
Comparative enrichment of the ANXA11 mutant proteins
in the nucleus suggests that mutations may impair
nuclear-cytoplasmic shuttling or result in a potential toxic
impairment of nuclear function.
ANXA11 mutants did not exert organism toxicity in
Drosophila but can only enhance the toxicity of human TDP43,
specifically, but not that of fly TBPH or human SOD1. Despite TBPH
being an ortholog of TDP43, the absence of enhancement of TBPH's
impact on lifespan and negative geotaxis by ANXA11 transgene
expression suggests a specific interaction between human A11 and
TDP43 that is not conserved with its Drosophila counterpart.
This could be attributed to differences in protein-protein
interactions, post-translational modification or cellular
localization between human TDP43 and Drosophila TBPH and may
be the reason why ANXA11 transgenes did not exert significant
toxicity in Drosophila.
The lack of interactions with SOD1 has implications
for ALS heterogeneity at a genetic and molecular level. ALS is a
multifactorial disease with diverse genetic contributors (1). The absence of a genetic link between
these two ALS-associated genes confirms distinct pathogenic
pathways for SOD1-associated ALS.
This also has implications for the development of
ALS models and the interpretation of previous studies (24-26):
It indicates that models based on overexpression or mutation of
ANXA11 may not be suitable for studying mechanisms related
to SOD1G85R pathology. This is crucial for ensuring the
accuracy and relevance of research models to specific subtypes of
ALS.
Thus, the present study demonstrated a novel and
potentially deleterious interaction between ANXA11 and TDP43 in ALS
pathology. The redistribution of TDP43 subcellular localization
suggested a potential regulatory role of ANXA11 in the
cellular trafficking or localization of TDP43, which could be
critical in understanding the pathological mechanisms of ALS. The
modulation of TDP43 localization by ANXA11 and the
mutation-dependent nature of this interaction opens novel avenues
for exploring the molecular basis of ALS. It suggests that
alterations in protein trafficking and localization are a key
aspect of disease progression as a result of nuclear pathology in
ANXA11-associated ALS and other types of neurodegenerative diseases
(28,29).
The N-terminus of ANX A11 binds and traffics RNA
granules with mutations, impairing LLPS (10,21).
ANXA11 mutations may potentially impair nuclear RNA dynamics and
TDP43 function at an early stage of the disease process (30). In post-mortem tissue staining of
ANXA11 mutant cases, partial axonal co-localisation of
phosphorylated TDP43 and immunoreactive ANXA11 has been observed
(5,12). Furthermore, patients with
multisystem proteinopathy harbouring a D40Y mutation exhibit
ANXA11/TDP43 cytoplasmic co-localisation in muscle (18). To the best of our knowledge the
association between endogenous nuclear ANXA11 (or N and C terminal
mutants) and TDP43 in neurons has yet to be established.
It is presently unclear whether the specificity of
the ANXA11-TDP43 axis is a fundamental molecular and mechanistic
difference between fly TBPH and human TDP43 or lack of full
conservation between human ANXA11 and fly ANXB11 in terms of their
association with TDP43 molecules. This is a limitation of the
present study, which is also limited by the use of exogenous
transgenes overexpressing the protein of interests. A knock in
model for ANAX11 and or TDP43/TBPH is required when attempting to
use Drosophila as a model for ALS genes whose toxicity is
associated with TDP43, such as ANXA11. It is essential to study the
ANXA11-TDP43 axis in other organisms to validate if the
relationship is preserved or if this is a human-specific factor.
Furthermore, understanding how specific ANXA11 mutations affect
TDP43 localization in human neurons and other model organisms could
provide insight into the heterogeneity of ALS symptoms and
progression.
Supplementary Material
ANXA11 has no genetic interaction with
SOD1G85R. (A) Survival and (B) negative geotaxis of
fruit flies overexpressing SOD1G85R were not impacted by
the presence of ANXA11. Annexin (ANX), Super Oxide Dismutase 1
(SOD1), Wild-Type (WT).
Acknowledgements
The authors would like to thank Dr Jemeen
Sreedharan, Dr Joe Bateman and Dr Frank Hirth (all from King's
College London, London, UK) for fly stocks and reagents.
Funding
Funding: The present study was supported by the UK Motor Neuron
Disease Association (grant no. 855-791).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JB, RH, DM and MY performed experiments. JB, RH, BS
and MF designed the experiments. MF and JB wrote the manuscript.
JB, RH and MF confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Casterton RL, Hunt RJ and Fanto M:
Pathomechanism heterogeneity in the amyotrophic lateral sclerosis
and frontotemporal dementia disease spectrum: Providing focus
through the lens of autophagy. J Mol Biol. 432:2692–2713.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kiernan MC, Vucic S, Cheah BC, Turner MR,
Eisen A, Hardiman O, Burrell JR and Zoing MC: Amyotrophic lateral
sclerosis. Lancet. 377:942–955. 2011.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mejzini R, Flynn LL, Pitout IL, Fletcher
S, Wilton SD and Akkari PA: ALS genetics, mechanisms, and
therapeutics: Where are we now? Front Neurosci.
13(1310)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Suzuki N, Nishiyama A, Warita H and Aoki
M: Genetics of amyotrophic lateral sclerosis: Seeking therapeutic
targets in the era of gene therapy. J Hum Genet. 68:131–152.
2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Smith BN, Topp SD, Fallini C, Shibata H,
Chen HJ, Troakes C, King A, Ticozzi N, Kenna KP, Soragia-Gkazi A,
et al: Mutations in the vesicular trafficking protein annexin A11
are associated with amyotrophic lateral sclerosis. Sci Transl Med.
9(eaad9157)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jiang Q, Lin J, Wei Q, Li C, Hou Y, Cao B,
Zhang L, Ou R, Liu K, Yang T, et al: Genetic analysis of and
clinical characteristics associated with ANXA11 variants in a
Chinese cohort with amyotrophic lateral sclerosis. Neurobiol Dis.
175(105907)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Johari M, Papadimas G, Papadopoulos C,
Xirou S, Kanavaki A, Chrysanthou-Piterou M, Rusanen S, Savarese M,
Hackman P and Udd B: Adult-onset dominant muscular dystrophy in
Greek families caused by Annexin A11. Ann Clin Transl Neurol.
9:1660–1667. 2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liu X, Wu C, He J, Zhang N and Fan D: Two
rare variants of the ANXA11 gene identified in Chinese patients
with amyotrophic lateral sclerosis. Neurobiol Aging. 74:235 e9–235
e12. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nagy ZF, Pal M, Salamon A, Kafui Esi
Zodanu G, Füstös D, Klivényi P and Széll M: Re-analysis of the
Hungarian amyotrophic lateral sclerosis population and evaluation
of novel ALS genetic risk variants. Neurobiol Aging. 116:1–11.
2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Nahm M, Lim SM, Kim YE, Park J, Noh MY,
Lee S, Roh JE, Hwang SM, Park CK, Kim YH, et al: ANXA11 mutations
in ALS cause dysregulation of calcium homeostasis and stress
granule dynamics. Sci Transl Med. 12(eaax3993)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nel M, Mahungu AC, Monnakgotla N, Botha
GR, Mulder NJ, Wu G, Rampersaud E, van Blitterswijk M, Wuu J,
Cooley A, et al: Revealing the mutational spectrum in Southern
Africans with amyotrophic lateral sclerosis. Neurol Genet.
8(e654)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sainouchi M, Hatano Y, Tada M, Ishihara T,
Ando S, Kato T, Tokunaga J, Ito G, Miyahara H, Toyoshima Y, et al:
A novel splicing variant of ANXA11 in a patient with amyotrophic
lateral sclerosis: Histologic and biochemical features. Acta
Neuropathol Commun. 9(106)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Teyssou E, Muratet F, Amador MD, Ferrien
M, Lautrette G, Machat S, Boillée S, Larmonier T, Saker S, Leguern
E, et al: Genetic screening of ANXA11 revealed novel mutations
linked to amyotrophic lateral sclerosis. Neurobiol Aging. 99:102
e11–102 e20. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang Y, Duan X, Zhou X, Wang R, Zhang X,
Cao Z, Wang X, Zhou Z, Sun Y and Peng D: ANXA11 mutations are
associated with amyotrophic lateral sclerosis-frontotemporal
dementia. Front Neurol. 13(886887)2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yang X, Sun X, Liu Q, Liu L, Li J, Cai Z,
Zhang K, Liu S, He D, Shen D, et al: Mutation spectrum of chinese
amyotrophic lateral sclerosis patients with frontotemporal
dementia. Orphanet J Rare Dis. 17(404)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang K, Liu Q, Liu K, Shen D, Tai H, Shu
S, Ding Q, Fu H, Liu S, Wang Z, et al: ANXA11 mutations prevail in
Chinese ALS patients with and without cognitive dementia. Neurol
Genet. 4(e237)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Natera-de Benito D, Olival J, Garcia-Cabau
C, Jou C, Roldan M, Codina A, Expósito-Escudero J, Batlle C,
Carrera-García L, Ortez C, et al: Common pathophysiology for ANXA11
disorders caused by aspartate 40 variants. Ann Clin Transl Neurol.
10:408–425. 2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Leoni TB, Gonzalez-Salazar C, Rezende TJR,
Hernández ALC, Mattos AHB, Coimbra Neto AR, da Graça FF, Gonçalves
JPN, Martinez ARM, Taniguti L, et al: A novel multisystem
proteinopathy caused by a missense ANXA11 Variant. Ann Neurol.
90:239–252. 2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kim EJ, Moon SY, Kim HJ, Jung NY, Lee SM
and Kim YE: Semantic variant primary progressive aphasia with a
pathogenic variant p.Asp40Gly in the ANXA11 gene. Eur J Neurol.
29:3124–3126. 2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fernandopulle M, Wang G, Nixon-Abell J,
Qamar S, Balaji V, Morihara R and St George-Hyslop PH: Inherited
and sporadic amyotrophic lateral sclerosis and fronto-temporal
lobar degenerations arising from pathological condensates of phase
separating proteins. Hum Mol Genet. 28 (R2):R187–R196.
2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liao YC, Fernandopulle MS, Wang G, Choi H,
Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, et al:
RNA granules hitchhike on lysosomes for long-distance transport,
using annexin A11 as a molecular tether. Cell. 179:147–164 e20.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sung W, Nahm M, Lim SM, Noh MY, Lee S,
Hwang SM, Kim YH, Park J, Oh KW, Ki CS, et al: Clinical and genetic
characteristics of amyotrophic lateral sclerosis patients with
ANXA11 variants. Brain Commun. 4(fcac299)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Casci I and Pandey UB: A fruitful
endeavor: Modeling ALS in the fruit fly. Brain Res. 1607:47–74.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sreedharan J, Neukomm LJ, Brown RH Jr and
Freeman MR: Age-Dependent TDP43-mediated motor neuron degeneration
requires GSK3, hat-trick, and xmas-2. Curr Biol. 25:2130–2136.
2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Watson MR, Lagow RD, Xu K, Zhang B and
Bonini NM: A Drosophila model for amyotrophic lateral
sclerosis reveals motor neuron damage by human SOD1. J Biol Chem.
283:24972–24981. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Romano G, Klima R, Buratti E, Verstreken
P, Baralle FE and Feiguin F: Chronological requirements of TDP43
function in synaptic organization and locomotive control. Neurobiol
Dis. 71:95–109. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mazaud D, Kottler B, Goncalves-Pimentel C,
Proelss S, Tüchler N, Deneubourg C, Yuasa Y, Diebold C, Jungbluth
H, Lai EC, et al: Transcriptional Regulation of the
Glutamate/GABA/Glutamine Cycle in adult glia controls motor
activity and seizures in Drosophila. J Neurosci.
39:5269–5283. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Marchica V, Biasetti L, Barnard J, Li S,
Nikolaou N, Frosch MP, Lucente DE, Eldaief M, King A, Fanto M, et
al: Annexin A11 mutations are associated with nuclear envelope
dysfunction in vivo and in human tissue. Brain: Jul 11, 2024 (Epub
ahead of print). doi: 10.1093/brain/awae226.
|
|
29
|
Baron O, Boudi A, Dias C, Schilling M,
Nölle A, Vizcay-Barrena G, Rattray I, Jungbluth H, Scheper W, Fleck
RA, et al: Stall in canonical autophagy-lysosome pathways prompts
nucleophagy-based nuclear breakdown in neurodegeneration. Curr
Biol. 27:3626–3642.e6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang A, Conicella AE, Schmidt HB, Martin
EW, Rhoads SN, Reeb AN, Nourse A, Ramirez Montero D, Ryan VH,
Rohatgi R, et al: A single N-terminal phosphomimic disrupts TDP43
polymerization, phase separation, and RNA splicing. EMBO J.
37(e97452)2018.PubMed/NCBI View Article : Google Scholar
|














