Introduction
Malignant pleural mesothelioma (MPM) is an
aggressive inflammatory cancer, predominantly arising from prior
exposure to asbestos (1). An
extended lag period between exposure and disease onset coupled with
the non-descript nature of the symptoms associated with this
disease means that the vast majority of patients when diagnosed
present at an advanced stage. Overall survival for MPM is poor with
most patients dying within one year of diagnosis (2,3). A
conservative estimate has suggested that globally ~43,000 people
die from this disease annually, but the actual number is probably
much greater (4,5). Although there has been some recent
advances in this disease, current standard of care is a combination
of pemetrexed and cisplatin chemotherapy (6), which is non-curative and results in a
response rate of ~40% (7).
Consequently, there is an urgent need to identify novel therapeutic
avenues in this disease to improve patient outcomes.
Epigenetics is often described as ‘chromatin-based
events that regulate DNA-templated processes’ (8). Furthermore, it is also now well
established that aberrant epigenetics are a common feature in
cancer, including mesothelioma (9).
Epigenetic regulation of chromatin is often
described as a code (10)
involving regulatory proteins that act as ‘readers’, ‘writers’ or
‘erasers’ of this code. The majority of these proteins utilize
various post-translational modifications such as histone
acetylation on core histones to elicit responses (11). The enzymes which write/erase
histone acetylation are called lysine acetyltransferases (KATs) and
histone deacetylases (HDACs), respectively (12). Since their discovery, significant
effort has been expended by the pharmaceutical sector to develop
agents that target these proteins. One such compound, vorinostat,
is a histone deacetylase inhibitor FDA approved for the treatment
of cutaneous T-cell lymphoma (13), was tested in a phase III clinical
trial as a second or third-line treatment in MPM, but unfortunately
did not improve overall survival and cannot be recommended as a
therapy for patients with MPM (14).
Lysine acetyltransferases are also emerging as
candidate therapeutic targets in cancer with the recent development
of several inhibitors targeting these proteins (15,16).
There are, however, a significant number of KATs and the KAT
superfamily itself can be separated into several subfamilies
(17). One of these is the MYST
family comprising KAT5-KAT8 (18).
MYST KATs have diverse functions affecting the majority of cellular
processes, ranging from gene regulation, cell cycle, stem cell
homeostasis and development and critically DNA damage repair
(18). A member of the MYST
family, KAT5 (also known as Tip60) is the catalytic subunit of the
NuA4 histone acetyltransferase complex which is involved in
transcriptional activation of select genes. This complex may be
required for the activation of transcriptional programs associated
with oncogene and proto-oncogene mediated growth induction, tumor
suppressor mediated growth arrest and replicative senescence,
apoptosis and DNA repair (19,20).
Critically, KAT5 has now also been linked to: i) activation of
excision repair cross-complementation group 1 (ERCC1) a critical
DNA damage repair protein in response to exposure to cisplatin
(21); and ii) with the
development of cisplatin resistance (22). As such, given that the standard
first-line therapy for MPM is cisplatin based (6), we examined whether the expression of
KAT5 is altered in MPM and if this protein could represent a novel
candidate target for intervention in this disease. Our results show
that KAT5 is significantly elevated in MPM, and targeting this
protein with small molecule inhibitors results in insignificant
anti-proliferative and apoptotic responses in MPM cell lines,
further underlining the therapeutic potential of targeting this
protein in mesothelioma.
Materials and methods
Primary tumor samples
Surgical specimens were obtained as discarded tumor
samples from patients who had undergone extended
pleuro-pneumonectomy at Glenfield Hospital (Leicester, UK). Benign
specimens were acquired from patients never diagnosed with MPM.
Informed consent was obtained from each patient, and the study was
conducted after formal approval from the relevant Hospital Ethics
Committee. Samples consisted of the following: 5 benign lesions and
17 MPM samples (epithelioid, n=7; biphasic, n=6; and sarcomatoid,
n=4).
Cell culture
All MPM cell lines were maintained in a humidified
atmosphere containing 5% CO2 in appropriate media
supplemented with 10% fetal bovine serum (FBS) and penicillin
streptomycin (500 U/ml). Cell culture reagents were purchased from
Lonza (Walkersville, MD, USA). Twenty-four MPM cell lines were used
in the study: LP9, Met5A, NCI-H2596, MMP, MMB, NCI-H2052, NCI-H28,
Ju77, One58, RS-5, DM-3, ACC-MESO-1, ACC-MESO-4, Y-MESO-8D,
Y-MESO-9, Y-MESO-12, Y-MESO-14, M14K, M24K, M25K, M28K, M33K, M38K
and LO68.
LP9, MMP and MMB were a generous gift from Dr Warren
Thomas (Royal College of Surgeons in Ireland, Dublin, Ireland).
ACC-MESO-1, ACC-MESO-4, Y-MESO-8D, Y-MESO-9, Y-MESO-12 and
Y-MESO-14 were generously provided by Yoshitaka Sekido (Aichi
Cancer Center Research Institute, Nagoya, Japan). NCI-H2052, One58
and JU77 cells were provided by Duncan Stewart (University of
Leicester, Leicester, UK). The NCI-H2596 and REN cell lines were
kindly provided by Dean Fennell (Queen's University, Belfast,
Northern Ireland). M14K, M24K, M25K, M28K, M33K and M38K were
generously provided by Dr Hannu Norppa (Finnish Institute of
Occupational Health, Helsinki, Finland). NCI-H28, and the
immortalized non-tumorigenic mesothelial cell line, Met-5A were
purchased from the ATCC (LGC Promochem, Teddington, UK). RS-5 and
DM-3 were purchased from the DSMZ (Leibniz Institute DSMZ-German
Collection of Microorganisms and Cell Cultures, Braunschweig,
Germany).
Reagents
MG 149 (23) was
purchased from Axon Medchem BV (Groningen, The Netherlands), and
dissolved in dimethyl sulfoxide (DMSO). Cells were serum starved
(0.5% FBS) for 24 h prior to the addition of drugs.
Proliferation assay
Cell proliferation was measured using a Cell
Proliferation BrdU ELISA (Roche Diagnostics Ltd., West Sussex, UK),
according to manufacturer's instructions. Briefly, cells (REN or
H26) were seeded at 3×103/well in a 96-well plate.
Following overnight incubation, cells were treated for 24 h with MG
149 at 1, 5, 10, 15, 20 and 25 μM. Absorbance was measured on a
plate reader at 450 nm with a reference wavelength set to 690 nm.
Untreated wells were used for normalization purposes and set to
100%.
Cellular apoptosis (FACS)
NCI-H226 cells were seeded in 6-well plates at a
density of 1×105 cells/well and were allowed to adhere
overnight. Subsequently the complete media was removed and the
cells washed with 100 ml PBS. Serum depleted media (0.5% FBS) was
added and the cells incubated for a further 24 h, then treated with
appropriate concentrations of drug, diluted in cell culture media,
for a further 48 h. Where appropriate, control cells were treated
with either vehicle or left untreated with media only. Following
treatment, culture media was removed, transferred to labeled FACS
tubes and placed on ice. Adhered cells were then trypsinised and
transferred to corresponding FACS tubes. Cells were pelleted by
centrifugation at 1,300 rpm for 3 min and the supernatant removed.
The cells were washed in 1 ml of 1X binding buffer (BB) diluted in
ice cold PBS, pelleted by centrifugation and resuspended in 100 μl
BB. Annexin V (2 μl) (IQ Products, Groningen, The Netherlands) was
added to each tube, with the exception of the negative control and
media only samples, and cells were incubated at 4°C for 20 min,
protected from light. Cells were again washed in 1 ml 1X binding
buffer and supernatant removed. Immediately before analysis by flow
cytometry, cells were resuspended in 400 μl BB containing 0.5 μg/ml
PI (Invitrogen, Paisley, UK), except the negative control and FMO
(fluorescence minus one) control for PI for which BB alone was
used.
Caspase-3/7 activation
Activation of caspase-3/7 was measured using a
FluoroFire Caspase-3/7 fluorescent assay kit according to the
manufacturer's instructions (Molecutools, Dublin, Ireland).
NCI-H226 cells were seeded in 96-well plates at a density of
4×103 cells/well and were allowed to adhere overnight.
Subsequently the complete media was removed and the cells washed
with 100 ml PBS. Serum depleted media (0.5% FBS) was added and the
cells incubated for a further 24 h, then treated with appropriate
concentrations of drug, diluted in cell culture media, for a
further 48 h.
RNA isolation and RT-PCR
amplification
Total RNA was extracted using TRI
reagent® (Molecular Research Center, Cincinnati, OH,
USA) according to manufacturer's instructions. Prior to First
Strand cDNA Synthesis, 250 ng (primary tumors) or 500 ng (cell
lines) total RNA was pre-treated by digestion with Amplification
grade DNase I (Sigma-Aldrich, St. Louis, MO, USA) according to the
manufacturer's instructions. cDNA was subsequently generated using
All-in-One cDNA Synthesis SuperMix (Biotool, Houston, TX, USA)
according to the manufacturer's instructions. Cell lines were
examined for the expression of KAT5 variants (RefSeq
NM_182710.2, NM_006388.3, NM_182709.2 and NM_001206833.1) and
18s rRNA by RT-PCR, using the following primers, or as
previously published (24).
KAT5v1, 5′-atggcggaggtggtgagtc-3; KAT5v2,
5′-gcggaggtgggggagataat-3′; and KAT5R,
5′-gaaaccacctccaccttccg-3′.
Amplification with KAT5v1 and KAT5R recognizes
variants 1 and 4, while amplification with KAT5v2 and KAT5R will
amplify variants 2 and 3.
PCR cycling conditions were 95°C for 5 min followed
by 35 cycles of 1 min at 95°C, 1 min at 58°C, 1 min at 72°C, for 35
cycles, with a final extension of 72°C for 10 min. A total of 8 μl
of the experimental RT-PCR product and 2 μl of the 18s rRNA RT-PCR
product was loaded and run on 2% agarose gel. Following image
capture, product quantification was performed using TINA 2.09c
(Raytest, Isotopenmeßgeräte GmbH, Straubenhardt, Germany)
densitometry software. The mRNA expression was normalized to the
loading control (18s rRNA), and was expressed as a ratio of target
mRNA expression: loading control expression.
Analysis of mRNA expression by
RT-qPCR
Validation of RT-PCR results was subsequently
confirmed using SYBR-Green based quantitative real-time PCR
(RT-qPCR). RT-qPCR reactions were carried out for CXCL8 using
previously published primers (24). Primers used to analyze CXCL1, and
CXCL13 were purchased from Real Time Primers, LLC (Elkins Park, PA,
USA).
RT-qPCRs were conducted on an Illumina Eco qPCR
using GoTaq® qPCR Master Mix (Promega) using a 2-step
qPCR program with the following cycling parameters as recommended
by Real Time Primers: An initial polymerase activation of 95°C for
2 min followed by 50 cycles of 95°C for 10 sec and
annealing/amplification at 58°C for 45 sec. A melting curve
analysis was conducted at the end of each PCR using 95°C for 15
sec, 55°C for 15 sec and a final 95°C for 15 sec. Data were
analysed using the default in-built ΔΔCq analysis settings for
relative quantification in Eco software.
Statistical analysis
All data are expressed as mean ± SEM unless stated
otherwise. Statistical analysis was performed with GraphPad Prism
v5.01 (GraphPad Software, La Jolla, CA, USA) using either unpaired
two-tailed Student's t-test or Mann-Whitney Student's t-test.
One-way analysis of variance (ANOVA) was used where groups in the
experiment were three or more. Following ANOVA, post-test analyses
utilized either the Tukey multiple comparisons test, or the
Dunnett's multiple comparison tests.
Results
Expression of KAT5 in primary
mesothelioma specimens
To assess the expression of KAT5 in a panel of
fresh-frozen mesothelioma samples with a cohort of benign pleura
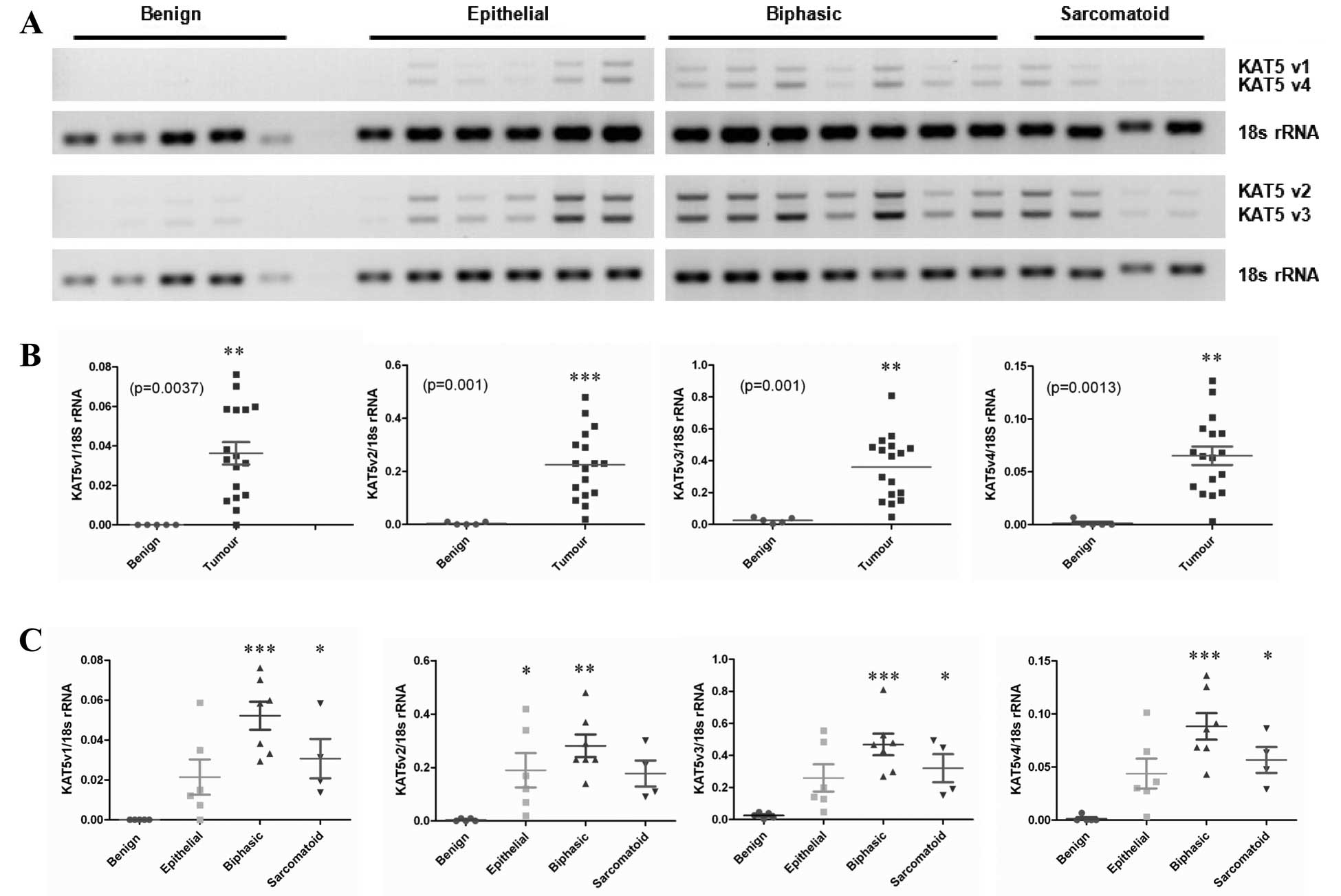
RT-PCR was performed (Fig. 1A).
KAT5 has four known RefSeq variants, and primers were designed to
distinguish between all four variants. Densitometric analysis of
the RT-PCR products revealed significantly increased expression of
all four variants in primary MPM tumor samples compared with normal
pleura (Fig. 1B). When stratified
according to histological subtype, the most predominant
upregulation was observed in the biphasic subtype (Fig. 1C).
Expression of KAT5 in a panel of normal
and malignant mesothelial cell lines
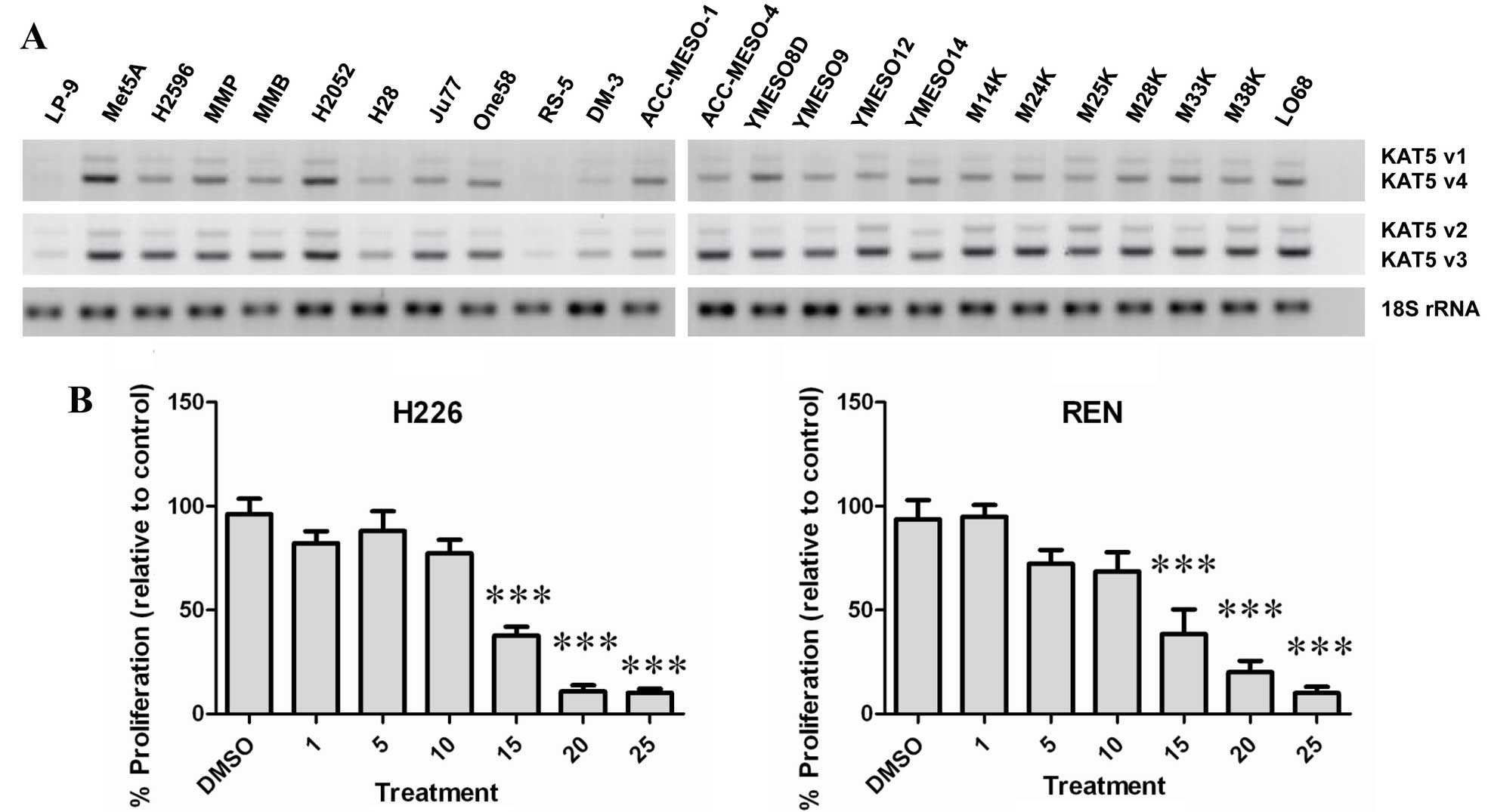
Utilizing RT-PCR, the expression of KAT5 was
examined in a panel of normal and malignant mesothelioma cell lines
(Fig. 2A). All cell lines tested
expressed varying levels of KAT5, with higher basal expression
observed predominantly in the mesothelioma lines, whereas low basal
levels of KAT5 were observed in the normal peritoneal mesothelial
cell line LP9 (Fig. 2A).
Inhibition of KAT5 leads to reduced
cellular proliferation
A small molecule inhibitor of KAT5 (MG 149) has been
developed (23). The effect of
this compound was subsequently tested on two cell lines (REN and
NCI-H226). At concentrations >10 μM, MG 149 was found to
significantly reduce the proliferative rate of both MPM cell types
(Fig. 2B).
Inhibition of KAT5 leads to increased
cellular apoptosis
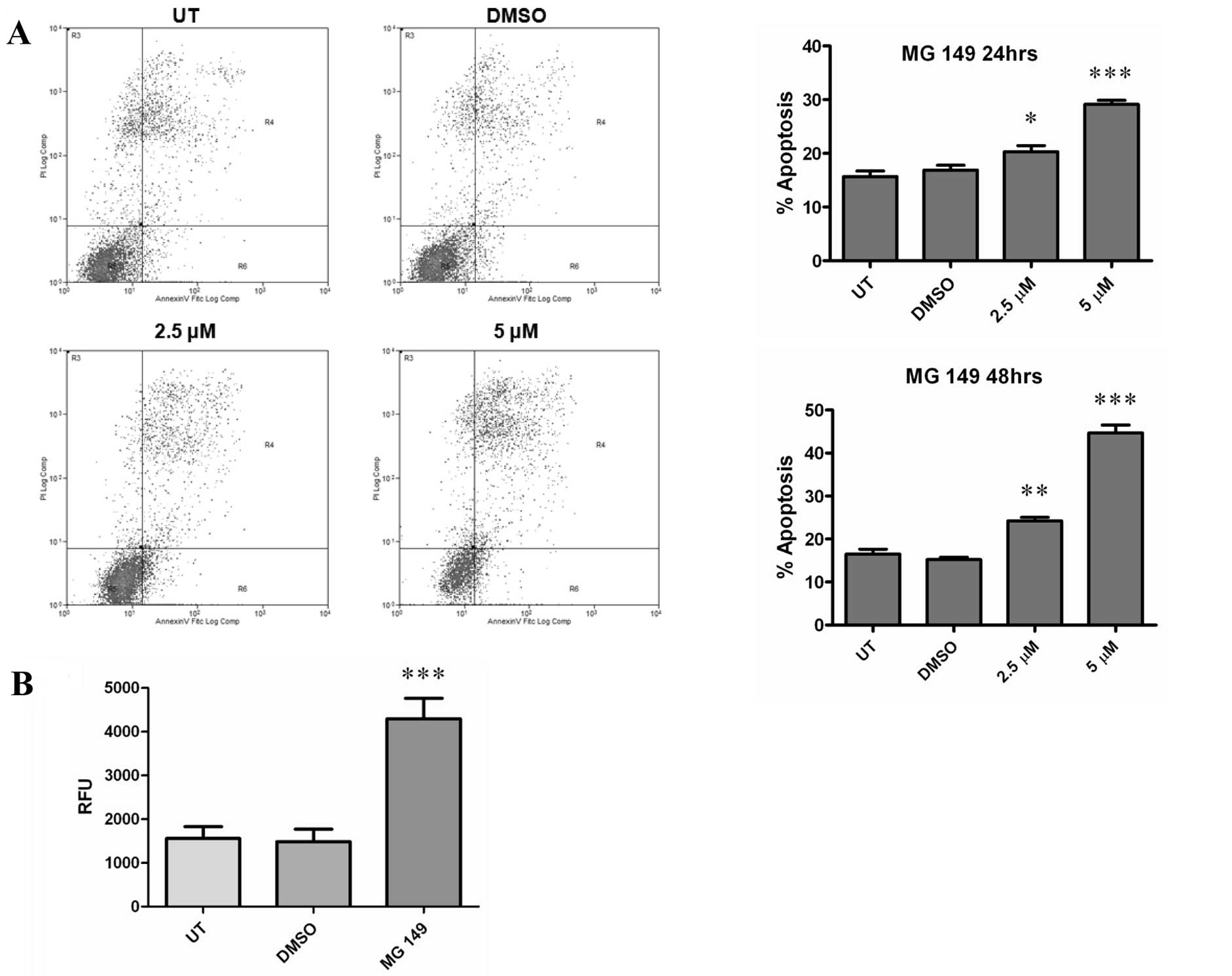
As cellular proliferation and viability were
affected by treatments with MG 149 we subsequently examined the
effects of this compound with respect to cellular apoptosis. Using
an Annexin V/FITC based FACs assay, 2.5 or 5 μM of MG 149 was found
to significantly increase cellular apoptosis at both 24 and 48 h
following treatment (Fig. 3A), and
was associated with increased caspase-3/7 activation (Fig. 3B).
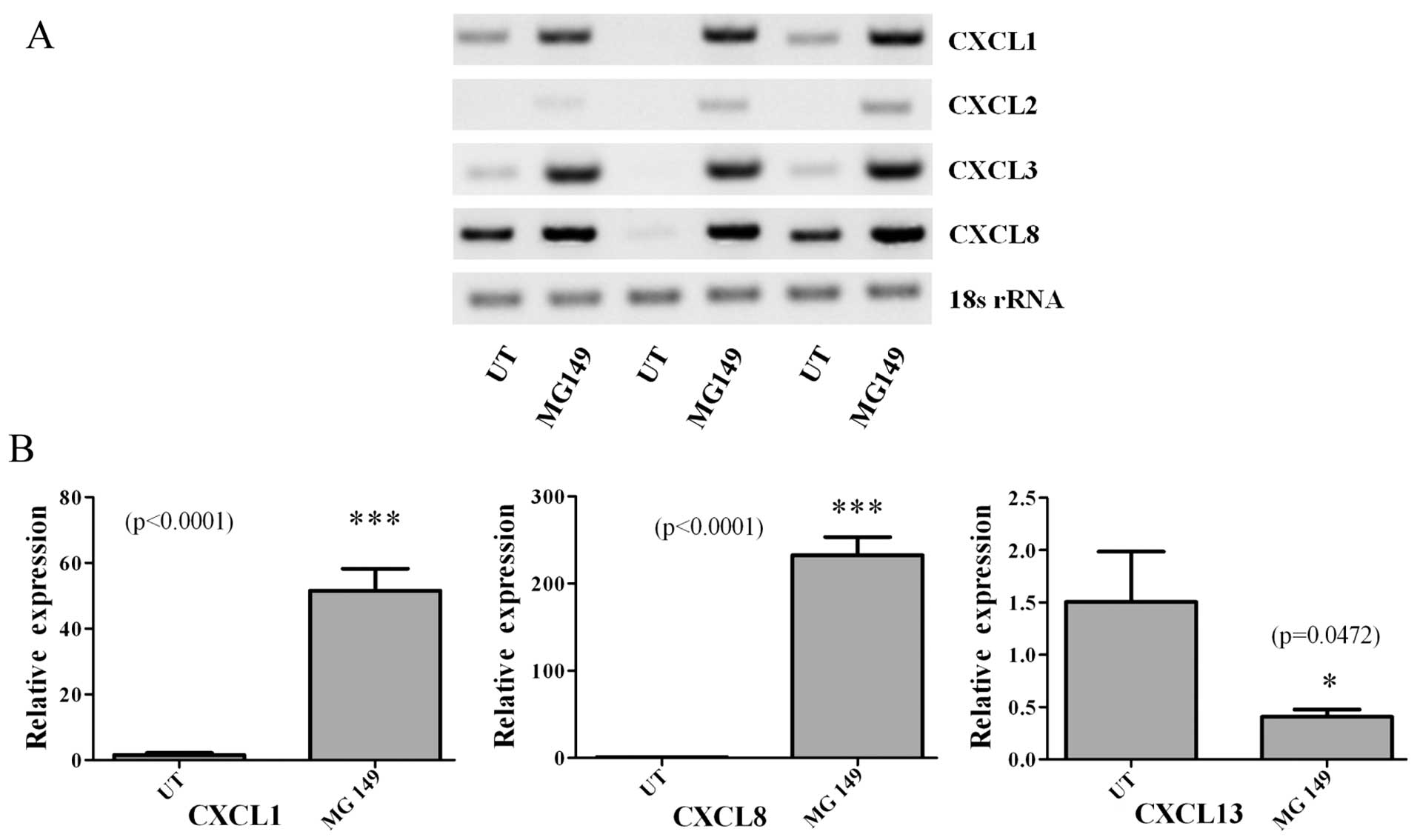
MG 149 treatments induce pro-inflammatory
cytokine expression
Given the effects of MG 149 on cellular
proliferation and apoptosis, and the fact that it targets an
epigenetic regulatory protein, we then examined the effects of MG
149 on the expression of members of the CXC (ELR+)
family namely CXCL1-3/GROα-γ, CXCL8/IL-8 and CXCL13. These are
pro-inflammatory chemokines, which we have previously shown to be
epigenetically regulated in non-small cell lung cancer (24), and for which some have also been
shown to be responsive to asbestos exposure (25). When cells were treated with MG 149
(15 μM for 24 h), RT-PCR analysis showed significant
induction/upregulation of four of the chemokines examined (Fig. 4A). Subsequently, using quantitative
PCR (qPCR), we confirmed and validated the upregulation of CXCL1
and CXCL8, with a small yet significant downregulation of CXCL13
(Fig. 4B).
Discussion
In the UK, it is estimated that 91,000 deaths from
mesothelioma are predicted to occur from 1968 to 2050 with around
61,000 of these occurring from 2007 onwards (26). The prevailing first-line
chemotherapy regimen for mesothelioma remains cisplatin/pemetrexed
(or cisplatin/raltitrexed) (6).
Critically, only ~40% of patients respond to this regimen, and we
therefore need to identify new agents for the treatment of this
cancer.
KAT5 is a member of the lysine acetyltransferases, a
large superfamily of epigenetic regulatory proteins which play a
number of important cellular roles, and which are emerging as
candidate targets for cancer therapy (15). In this regard, KAT5 has been shown
to be associated with resistance to cisplatin based therapy in the
lung cancer setting (21,22), suggesting that its expression could
also be important in mesothelioma cancer. We therefore examined the
levels of this lysine acetyltransferase in a panel of primary
tumors compared to benign pleura. Our results clearly demonstrated
that expression of KAT5 mRNA was significantly elevated in the
tumors compared to normal pleura (Fig.
1), and was ubiquitously expressed in a panel of mesothelioma
cell lines (Fig. 2). In agreement
with our data from primary tumors the expression of KAT5 was less
highly expressed in the normal LP9 cell line compared to MPM cancer
cell lines (Fig. 2), suggesting
that KAT5 may be a potential therapeutic target in
mesothelioma.
Several KAT5 specific inhibitors have been developed
including MG 149 (23), TH1834
(27), Nu9056 (28) and various H3-CoA inhibitors
(29). We exposed MPM cells to one
of these inhibitors, MG 149, and demonstrated that KAT5 inhibition
resulted in both significant anti-proliferative effects (Fig. 2B) and the induction of cellular
apoptosis (Fig. 3) in cells.
Epigenetic therapies have had mixed results within
the clinical setting particularly for agents such as histone
deacetylase inhibitors (HDACi) when used as a monotherapy (30). Prior to 2013, estimates suggest
that 490 clinical trials had been conducted with HDACi with very
limited success (31), and indeed
one of the most conspicuous failures for an HDACi was the
VANTAGE-014 trial in mesothelioma, which examined the effect of
vorinostat in patients with advanced malignant pleural mesothelioma
who had progressed on previous chemotherapy (14). Nevertheless, epigenetic therapies
remain attractive targets for the treatment of cancer, and recent
‘breakthrough therapy FDA designation’ for entinostat for breast
cancer when added to exemestane in postmenopausal women with
ER+ metastatic breast cancer whose cancer had progressed
after treatment with a non-steroidal aromatase inhibitor (32) and approval of panobinostat for the
treatment of multiple myeloma (33) suggest that a better understanding
of underlying disease and better stratification of patients may
improve clinical outcomes for epigenetic therapies.
MG 149 was shown to induce or upregulate the
expression of several pro-inflammatory chemokines/cytokines
including CXCL8/IL-8 (Fig. 4).
Induction of IL-8 by anti-cancer regimens has been seen before in
experimental models of mesothelioma (34). Levels of IL-8 have also been shown
to be increased in mesothelioma patients undergoing therapy
following pleurectomy or extrapleural pneumonectomy (EPP) (35), or in patients exposed to
intrapleural administration of tumor necrosis factor-alpha (TNF-α)
(36). However, elevation of IL-8
levels may not be a reliable marker for response to therapy
(37), and the induction of
pro-inflammatory cytokines/chemokine's by MG-149 suggests that as a
therapy it may elicit cytokine response syndrome (CRS) (38), sometimes observed in MPM (39). Indeed, an additional potential
caveat to the utility of MG 149 as a novel therapeutic modality in
MPM comes from early research which demonstrated that IL-8 is
pro-angiogenic and acts as an autocrine growth factor in MPM
(40,41). Furthermore, inhibition of IL-8 was
found to decrease mesothelioma growth both in vitro and
in vivo (42,43).
In conclusion, KAT5 may represent a novel
therapeutic target for the treatment of MPM. As its expression has
been linked to resistance to cisplatin, and current first-line
therapy of mesothelioma utilize platin based regimens, future
studies should examine whether KAT5 expression is linked to patient
outcome, and test the possibility that agents targeting this
protein could potentially sensitize (or resensitize) patients to
such regimens.
Acknowledgements
The authors are grateful to Drs Warren Thomas,
Yoshitaka Sekido, Dean Fennell and Hannu Norppa for their
generosity in providing access to various mesothelioma cell lines.
The present study was supported in part by funding for consumables
from the Masters in Translational Oncology program (TCD) for Sian
Cregan.
References
|
1
|
Wagner JC, Sleggs CA and Marchand P:
Diffuse pleural mesothelioma and asbestos exposure in the North
Western Cape Province. Br J Ind Med. 17:260–271. 1960.PubMed/NCBI
|
|
2
|
Peto J, Hodgson JT, Matthews FE and Jones
JR: Continuing increase in mesothelioma mortality in Britain.
Lancet. 345:535–539. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson BW, Musk AW and Lake RA:
Malignant mesothelioma. Lancet. 366:397–408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Driscoll T, Nelson DI, Steenland K, Leigh
J, Concha-Barrientos M, Fingerhut M and Prüss-Ustün A: The global
burden of disease due to occupational carcinogens. Am J Ind Med.
48:419–431. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park EK, Takahashi K, Hoshuyama T, Cheng
TJ, Delgermaa V, Le GV and Sorahan T: Global magnitude of reported
and unreported mesothelioma. Environ Health Perspect. 119:514–518.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baas P, Fennell D, Kerr KM, Van Schil PE,
Haas RL and Peters S: Malignant pleural mesothelioma: ESMO Clinical
Practice Guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 26(Suppl 5): v31–v39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S,
Manegold C, et al: Phase III study of pemetrexed in combination
with cisplatin versus cisplatin alone in patients with malignant
pleural mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baird A, Richard D, O'Byrne KJ and Gray
SG: Epigenetic therapy in lung cancer and mesothelioma. Epigenetic
Cancer Therapy. 1st edition. Gray SG: Academic Press; pp. 189–213.
2015, View Article : Google Scholar
|
|
10
|
Jenuwein T and Allis CD: Translating the
histone code. Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rothbart SB and Strahl BD: Interpreting
the language of histone and DNA modifications. Biochim Biophys
Acta. 1839:627–643. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Allis CD, Berger SL, Cote J, Dent S,
Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar
R, et al: New nomenclature for chromatin-modifying enzymes. Cell.
131:633–636. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mann BS, Johnson JR, Cohen MH, Justice R
and Pazdur R: FDA approval summary: Vorinostat for treatment of
advanced primary cutaneous T-cell lymphoma. Oncologist.
12:1247–1252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krug LM, Kindler HL, Calvert H, Manegold
C, Tsao AS, Fennell D, Öhman R, Plummer R, Eberhardt WE, Fukuoka K,
et al: Vorinostat in patients with advanced malignant pleural
mesothelioma who have progressed on previous chemotherapy
(VANTAGE-014): A phase 3, double-blind, randomised,
placebo-controlled trial. Lancet Oncol. 16:447–456. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Farria A, Li W and Dent SY: KATs in
cancer: Functions and therapies. Oncogene. 34:4901–4913. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Furdas SD, Kannan S, Sippl W and Jung M:
Small molecule inhibitors of histone acetyltransferases as
epigenetic tools and drug candidates. Arch Pharm (Weinheim).
345:7–21. 2012. View Article : Google Scholar
|
|
17
|
Yang XJ: The diverse superfamily of lysine
acetyltransferases and their roles in leukemia and other diseases.
Nucleic Acids Res. 32:959–976. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sapountzi V and Côté J: MYST-family
histone acetyltransferases: Beyond chromatin. Cell Mol Life Sci.
68:1147–1156. 2011. View Article : Google Scholar
|
|
19
|
Doyon Y and Côté J: The highly conserved
and multifunctional NuA4 HAT complex. Curr Opin Genet Dev.
14:147–154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bird AW, Yu DY, Pray-Grant MG, Qiu Q,
Harmon KE, Megee PC, Grant PA, Smith MM and Christman MF:
Acetylation of histone H4 by Esa1 is required for DNA double-strand
break repair. Nature. 419:411–415. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Den Broeck A, Nissou D, Brambilla E,
Eymin B and Gazzeri S: Activation of a Tip60/E2F1/ERCC1 network in
human lung adenocarcinoma cells exposed to cisplatin.
Carcinogenesis. 33:320–325. 2012. View Article : Google Scholar
|
|
22
|
Miyamoto N, Izumi H, Noguchi T, Nakajima
Y, Ohmiya Y, Shiota M, Kidani A, Tawara A and Kohno K: Tip60 is
regulated by circadian transcription factor clock and is involved
in cisplatin resistance. J Biol Chem. 283:18218–18226. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ghizzoni M, Wu J, Gao T, Haisma HJ, Dekker
FJ and George Zheng Y: 6-alkylsalicylates are selective Tip60
inhibitors and target the acetyl-CoA binding site. Eur J Med Chem.
47:337–344. 2012. View Article : Google Scholar :
|
|
24
|
Baird AM, Gray SG and O'Byrne KJ:
Epigenetics underpinning the regulation of the CXC
(ELR+) chemokines in non-small cell lung cancer. PLoS
One. 6:e145932011. View Article : Google Scholar
|
|
25
|
Dragon J, Thompson J, MacPherson M and
Shukla A: Differential susceptibility of human pleural and
peritoneal mesothelial cells to asbestos exposure. J Cell Biochem.
116:1540–1552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan E, Warren N, Darnton AJ and Hodgson
JT: Projection of mesothelioma mortality in Britain using Bayesian
methods. Br J Cancer. 103:430–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao C, Bourke E, Scobie M, Famme MA,
Koolmeister T, Helleday T, Eriksson LA, Lowndes NF and Brown JA:
Rational design and validation of a Tip60 histone acetyltransferase
inhibitor. Sci Rep. 4:53722014.PubMed/NCBI
|
|
28
|
Coffey K, Blackburn TJ, Cook S, Golding
BT, Griffin RJ, Hardcastle IR, Hewitt L, Huberman K, McNeill HV,
Newell DR, et al: Characterisation of a Tip60 specific inhibitor,
NU9056, in prostate cancer. PLoS One. 7:e455392012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang C, Ngo L and Zheng YG: Rational
design of substrate-based multivalent inhibitors of the histone
acetyltransferase Tip60. ChemMedChem. 9:537–541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gryder BE, Sodji QH and Oyelere AK:
Targeted cancer therapy: Giving histone deacetylase inhibitors all
they need to succeed. Future Med Chem. 4:505–524. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yardley DA, Ismail-Khan RR, Melichar B,
Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee
MJ and Trepel JB: Randomized phase II, double-blind,
placebo-controlled study of exemestane with or without entinostat
in postmenopausal women with locally recurrent or metastatic
estrogen receptor-positive breast cancer progressing on treatment
with a nonsteroidal aromatase inhibitor. J Clin Oncol.
31:2128–2135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fenichel MP: FDA approves new agent for
multiple myeloma. J Natl Cancer Inst. 107:djv1652015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lansley SM, Varano Della Vergiliana JF,
Cleaver AL, Ren SH, Segal A, Xu MY and Lee YC: A commercially
available preparation of Staphylococcus aureus bio-products
potently inhibits tumour growth in a murine model of mesothelioma.
Respirology. 19:1025–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yom SS, Busch TM, Friedberg JS, Wileyto
EP, Smith D, Glatstein E and Hahn SM: Elevated serum cytokine
levels in mesothelioma patients who have undergone pleurectomy or
extrapleural pneumonectomy and adjuvant intraoperative photodynamic
therapy. Photochem Photobiol. 78:75–81. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stam TC, Swaak AJ, Kruit WH, Stoter G and
Eggermont AM: Intrapleural administration of tumour necrosis
factor-alpha (TNFalpha) in patients with mesothelioma: Cytokine
patterns and acute-phase protein response. Eur J Clin Invest.
30:336–343. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nowak AK, Millward MJ, Creaney J, Francis
RJ, Dick IM, Hasani A, van der Schaaf A, Segal A, Musk AW and Byrne
MJ: A phase II study of intermittent sunitinib malate as
second-line therapy in progressive malignant pleural mesothelioma.
J Thorac Oncol. 7:1449–1456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee DW, Gardner R, Porter DL, Louis CU,
Ahmed N, Jensen M, Grupp SA and Mackall CL: Current concepts in the
diagnosis and management of cytokine release syndrome. Blood.
124:188–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nowak AK, Cook AM, McDonnell AM, Millward
MJ, Creaney J, Francis RJ, Hasani A, Segal A, Musk AW, Turlach BA,
et al: A phase 1b clinical trial of the CD40-activating antibody
CP-870,893 in combination with cisplatin and pemetrexed in
malignant pleural mesothelioma. Ann Oncol. 26:2483–2490.
2015.PubMed/NCBI
|
|
40
|
Antony VB, Hott JW, Godbey SW and Holm K:
Angiogenesis in mesotheliomas. Role of mesothelial cell derived
IL-8. Chest. 109(Suppl): 21S–22S. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Galffy G, Mohammed KA, Dowling PA, Nasreen
N, Ward MJ and Antony VB: Interleukin 8: An autocrine growth factor
for malignant mesothelioma. Cancer Res. 59:367–371. 1999.PubMed/NCBI
|
|
42
|
Cioce M, Canino C, Pulito C, Muti P,
Strano S and Blandino G: Butein impairs the protumorigenic activity
of malignant pleural mesothelioma cells. Cell Cycle. 11:132–140.
2012. View Article : Google Scholar
|
|
43
|
Galffy G, Mohammed KA, Nasreen N, Ward MJ
and Antony VB: Inhibition of interleukin-8 reduces human malignant
pleural mesothelioma propagation in nude mouse model. Oncol Res.
11:187–194. 1999.PubMed/NCBI
|