Introduction
The tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is a cytokine that belongs to the tumor necrosis
factor superfamily. TRAIL has emerged as a promising
cancer-selective anticancer drug. It inhibits cell proliferation
and induces cell death in a variety of cancer cell types while has
minimal cytotoxicity toward normal cells (1–4).
TRAIL primarily triggers the extrinsic and intrinsic apoptotic
pathways by binding to two death receptors (DRs), TRAIL receptor
(TRAIL-R)1/DR4 and TRAIL-R2/DR5 (5,6).
TRAIL has also been shown to trigger other cell death modalities
including autophagy (7,8) and necroptosis (9,10) in
some cancers. Different cancers including malignant melanoma (MM)
and osteosarcoma (OS) cells are resistant to TRAIL-induced
apoptosis (11–15). Accordingly, the combined use of a
certain drug that enables to alleviate drug resistance is essential
for effective TRAIL therapy of these cancers.
Ca2+ is an essential intracellular second
messenger whose level is tightly regulated. Finely and
spatiotemporally tuning of Ca2+ results in short and
synchronized Ca2+ waves, which are primarily required
for energy production, cell function and survival (16). On the other hand, a significant and
persistent increase in Ca2+ is a master cause of cell
death. An excessive rise in mitochondrial Ca2+
concentration ([Ca2+]mit) results in
increased permeability of the inner mitochondrial membrane. The
mitochondrial permeability transition (MPT) in turn leads to a
rapid collapse of mitochondrial membrane potential, loss of ATP,
osmotic rupture of the outer mitochondrial membrane. Ultimately,
the loss of ATP and the fall of the mitochondrial integrity lead to
necrosis (17,18). Mitochondrial Ca2+
overload also causes apoptosis. The rupture of the outer
mitochondrial membrane can result in the release of different
pro-apoptotic proteins such as cytochrome c and
apoptosis-inducing factor (19).
Recent evidence suggests that Ca2+ also plays a
regulatory role in other cell death modalities such as autophagy
and anoikis (20). Moreover,
different cancer cell types exhibit tumor-specific traits in
Ca2+ dynamics, which contribute to tumorigenesis,
malignant phenotypes, drug resistance, increased proliferation,
evasion from apoptosis and survival (21). Thus, Ca2+ is emerging as
a new target for cancer treatment (22,23).
Ca2+ dynamics in MM and OS and the role of
Ca2+ in TRAIL cytotoxicity toward these two cancers
remain largely unclear. Previously, we showed that acute TRAIL
treatment increased both [Ca2+]cyt and
[Ca2+]mit in several human MM and OS cell
lines (24). Unexpectedly, the
Ca2+ signals served as a pro-survival factor rather than
a pro-apoptotic factor, since overall Ca2+ removal or
specific removal of mitochondrial Ca2+ sensitized these
cells to TRAIL cytotoxicity. Our data suggested that the two cancer
cell types were resistant to mitochondrial Ca2+ overload
caused by TRAIL.
Mitochondria are dynamic organelles whose structure
is regulated by a mechanic mechanism encompassing fission and
fusion processes. Mitochondrial network homeostasis is critical for
cell function and survival (25)
since it is essential for maintaining mitochondrial functions such
as energy supply and metabolic activity. Mitochondrial dynamics is
regulated by dynamin-related proteins with GTPase activity.
Dynamin-related protein 1 (Drp1) regulates mitochondrial fission,
while mitofusin 1/2 and optic atrophy 1 control mitochondrial
fusion and cristae organization (26). It is widely accepted that cancer
cells alter mitochondrial dynamics to resist apoptosis and adapt
their nonphysiological bioenergetics microenvironments (27). Accordingly, disruption of the
mitochondrial dynamics may be a promising strategy to target
apoptosis-resistant cancer cells. Indeed, increasing body of
evidence indicates that inhibiting mitochondrial fission and fusion
regulators cause a severe mitochondrial dysfunction and apoptosis
in a variety of cancer cell types. However, the role of
mitochondrial fission in apoptosis is a matter of debate.
Mitochondrial fission exhibits inverse (pro-apoptotic or
anti-apoptotic) functions depending on the cell type and the
applied apoptotic stimuli (28–32).
Previously, we demonstrated that TRAIL evokes mitochondrial
fragmentation in a tumor-selective manner and that inhibition or
knockdown of Drp1 caused mitochondrial hyperfusion and sensitized
MM and OS cells to TRAIL-induced apoptosis (33,34).
These observations suggest that mitochondrial fission is a critical
pro-survival event in these cancer cells upon TRAIL treatment. In
this study, we investigated the mechanisms of the resistance to
mitochondrial Ca2+ overload with a particular interest
in the relevance to mitochondrial dynamics. Here we report that
mitochondrial Na+/Ca2+ exchanger (NCLX) plays
a pivotal role in the resistance to mitochondrial Ca2+
overload. Eventually, blockade of NCLX caused mitochondrial
Ca2+ overload and sensitized MM and OS cells to TRAIL
cytotoxicity. Moreover, we found that mitochondrial Ca2+
overload and removal regulated mitochondrial fission positively and
negatively, respectively while commonly exacerbated TRAIL-induced
mitochondrial network abnormalities, apoptosis, and non-apoptotic
cell death.
Materials and methods
Materials
Soluble recombinant human TRAIL was obtained from
Enzo Life Sciences (San Diego, CA, USA). Agonistic anti-human
TRAIL-R2/TNFRSF10B antibody (clone 71903 #MAB631-100) was purchased
from R&D Systems (Minneapolis, MN, USA). Antimycin A, FCCP, and
the pan-caspase-inhibitor z-VAD-fluoromethylketone (Z-VAD-FMK) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). All insoluble
reagents were dissolved in dimethyl-sulfoxide and diluted with high
glucose-containing Dulbecco's modified Eagle's medium
(Sigma-Aldrich) supplemented with 10% fetal bovine serum
(Sigma-Aldrich; FBS/DMEM) or Hank's balanced salt solution (HBSS)
(pH 7.4) to a final concentration of <0.1% before use.
Cell culture
Human MM (A375, A2058, SK-MEL-2) and OS (MG63,
SAOS-2, HOS) cell lines and WI-38 fibroblasts were obtained from
Health Science Research Resource Bank (Osaka, Japan). Human TE85
and 143B OS cells were kindly gifted by Dr T. Ando (Yamanashi
University). Human dermal fibroblasts (HDF) from facial dermis were
obtained from Cell Applications (San Diego, CA, USA). These cells
were cultured in FBS/DMEM supplemented with 100 U/ml penicillin and
100 µg streptomycin (Thermo Fisher Scientific, Rochester,
NY, USA) in a 5% CO2 incubator. Cells were harvested by
incubating with 0.25% trypsin-EDTA (Thermo Fisher Scientific) for 5
min at 37°C.
Ca2+ measurements
Changes in [Ca2+]cyt and
[Ca2+]mit levels were measured using Fluo
4-AM and rhod 2-AM (Dojindo Kumamoto, Japan), respectively as
previously described (24). For
improvement of mitochondrial localization of rhod 2-AM, it was
reduced to the colorless, nonfluorescent dihydrorhod 2-AM by sodium
borohydride, according to the manufacturer's protocol. Cells were
loaded with 4 µM each of Fluo 4-AM or dihydrorhod 2-AM for
40 min at 37°C, washed with HBSS. Then, the cells
(1×106/ml) were resuspended in HBSS in 96-well plates.
The cells were manually added with the agents to be tested. Then,
the cells were measured for fluorescence in a microplate reader
(Fluoroskan Ascent, Thermo Fisher Scientific) with excitation and
emission at 485 and 538 nm (for Fluo 4-AM) and 542 and 592 nm (for
rhod 2-AM), respectively.
Cell viability and apoptosis
measurements
Cell growth was measured by WST-8 assay using Cell
Counting Reagent SF (Nacalai Tesque, Kyoto, Japan) as previously
described (24). This method is a
colorimetric assay based on the formation of a water-soluble
formazan product. Briefly, cells (8×103/well) were
seeded in 96-well plates and cultured with the agents to be tested
for 72 h at 37°C in a 5% CO2 incubator. Then 1/10 volume
of WST-8 reagent was added, incubated for 1 h at 37°C and
absorbance at 450 nm was measured using a microplate reader (ARVO
MX, Perkin-Elmer Japan).
Caspase-3/7 activation, membrane
integrity, and cell death assay
Caspase-3/7 activation, membrane integrity, and cell
death were simultaneously measured by Muse™ Cell Analyzer (Merck
Millipore, Darmstadt, Germany) using Muse Caspase-3/7 kit as
previously described (24).
Briefly, cells (1×105/ml) in 24-well plates were treated
with the agents to be tested for 24 h in 10% FBS/DMEM at 37°C and
then stained with a novel caspase-3/7 reagent NucView™ and
7-amino-actinomycin D (7-AAD), a dead cell marker in the kit. 7-AAD
is excluded from healthy and early apoptotic cells, while permeates
late apoptotic and dead cells. Accordingly, four cell populations
can be distinguished by the kit; live cells,
caspase−/7-AAD−; early apoptotic cells,
caspase+/7-AAD−; late apoptotic/dead cells,
caspase+/7-AAD+; necrotic cells,
caspase−/7-AAD+.
Live-cell mitochondrial network
imaging
The mitochondrial network was analyzed as previously
described (34) with minor
modifications. Briefly, cells in FBS/DMEM (3×104/well)
adherent on 8-well chambered coverslips were treated with the
agents to be tested for 24 h at 37°C in a 5% CO2
incubator. After removing the medium by aspiration, the cells were
washed with fresh FBS/DMEM and stained with 20 nM MitoTracker Red
CMXRos for 1 h at 37°C in the dark in a 5% CO2
incubator. The cells were then washed with and immersed in
FluoroBrite™ DMEM (Thermo Fisher Scientific). Images were obtained
using a BZ X-700 Fluorescence Microscope (Keyence, Osaka, Japan)
equipped with a 100×, 1.40 n.a. UPlanSApo Super-Apochromat,
coverslip-corrected oil objective (Olympus, Tokyo, Japan). Images
were analyzed using BZ-H3A application software (Keyence) and free
NIH ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Data were analyzed by one-way analysis of variance
followed by the Tukey's post hoc test using an add-in software for
Excel 2016 for Windows (SSRI, Tokyo, Japan). All values were
expressed as means ± SD and P<0.05 was considered to be
significant.
Results
TRAIL-resistant tumor cells are highly
tolerant to mitochondrial Ca2+ overload by the drug
Previously, we demonstrated that TRAIL induces
[Ca2+]cyt and [Ca2+]mit
in several MM and OS cell lines in a dose-dependent manner
(24). However, we noticed that in
different cell lines the effects of TRAIL on
[Ca2+]cyt and [Ca2+]mit
were not always in parallel. Therefore, we studied the impact of
TRAIL on [Ca2+]cyt and
[Ca2+]mit in more detail. The cells were
loaded with Ca2+ probes, added with the agents to be
tested, and analyzed for their fluorescence in a microplate
fluorescence reader. This manual addition alone led to an immediate
and transient increase in [Ca2+]cyt, probably
owing to a mechanical stress-sensitive cation channel. After that,
[Ca2+]cyt returned to the baseline within 3–5
min. On the other hand, [Ca2+]cyt was
minimally changed for at least the initial 10 min when the cells
were allowed to stand without any addition of the materials.
Throughout this study, we monitored early Ca2+ responses
including this tentative mechanical [Ca2+]cyt
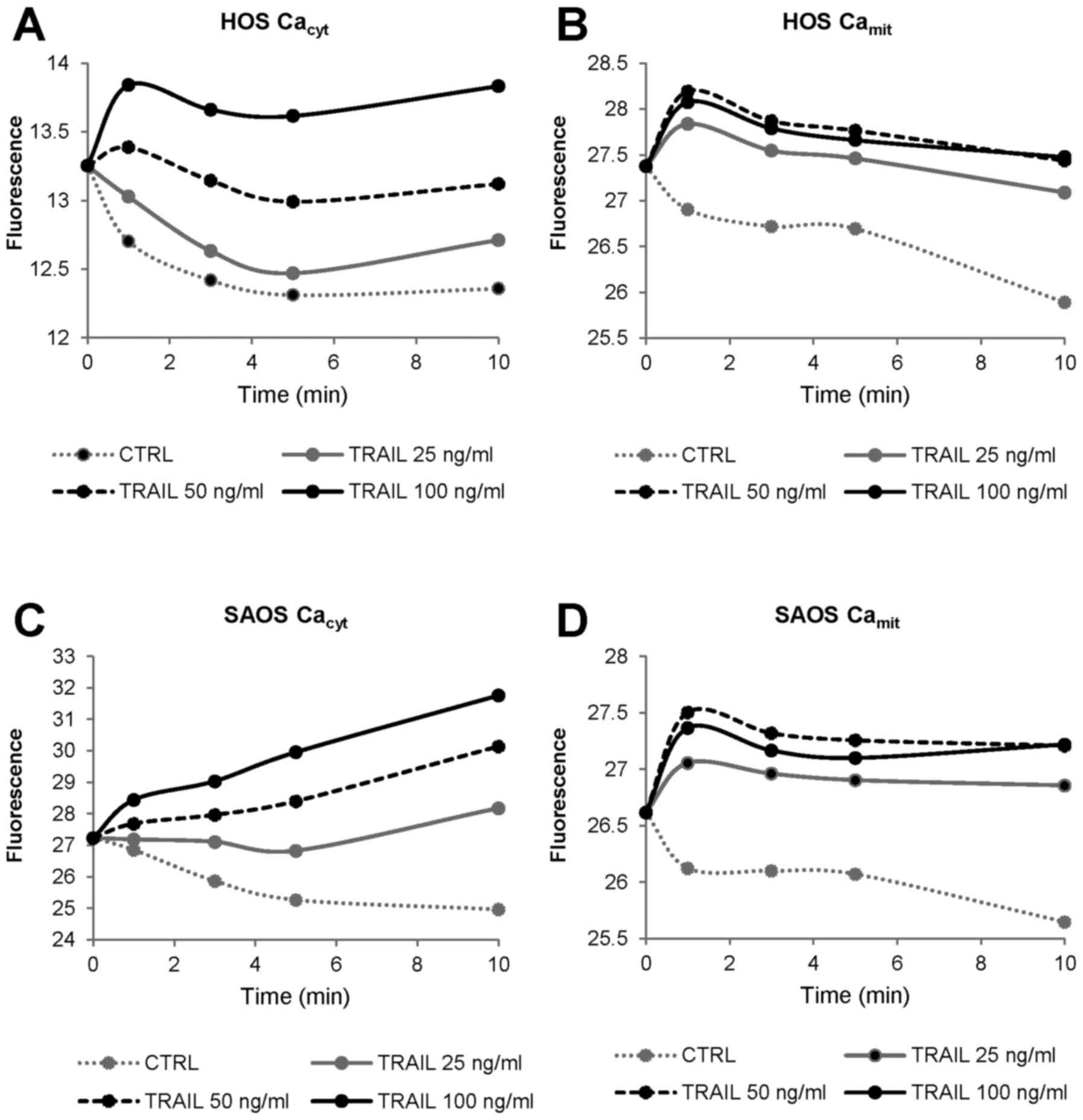
changes. Fig. 1 shows
representative results in HOS and SAOS-2 cells. TRAIL increased
[Ca2+]cyt in a dose-dependent manner with the
minimal effective dose of 25 ng/ml (Fig. 1A and C), while it increased
[Ca2+]mit maximally at 50 ng/ml (Fig. 1B and D). Strikingly, under the
conditions, the basal [Ca2+]mit in control
cells declined gradually over time. Whereas, relatively
TRAIL-sensitive cells including A375 cells, the basal
[Ca2+]mit was unchanged at least for 10 min,
and [Ca2+]mit elevated in parallel with
[Ca2+]cyt in response to TRAIL (data not
shown). Collectively, these findings suggest that TRAIL-resistant
tumor cells are highly tolerant to mitochondrial Ca2+
overload.
CGP-37157 causes mitochondrial
Ca2+ overload in MM and OS cells
NCLX plays a fundamental role in Ca2+
extrusion from the mitochondria in a variety of cell types
(35–37). Also, we previously observed that
treatment with CGP-37157, a specific inhibitor of NCLX, led to a
substantial increase in [Ca2+]mit in OS cells
(24). Therefore, we hypothesized
that increased Ca2+ efflux through NCLX might contribute
to the tolerance of mitochondrial Ca2+ overload in
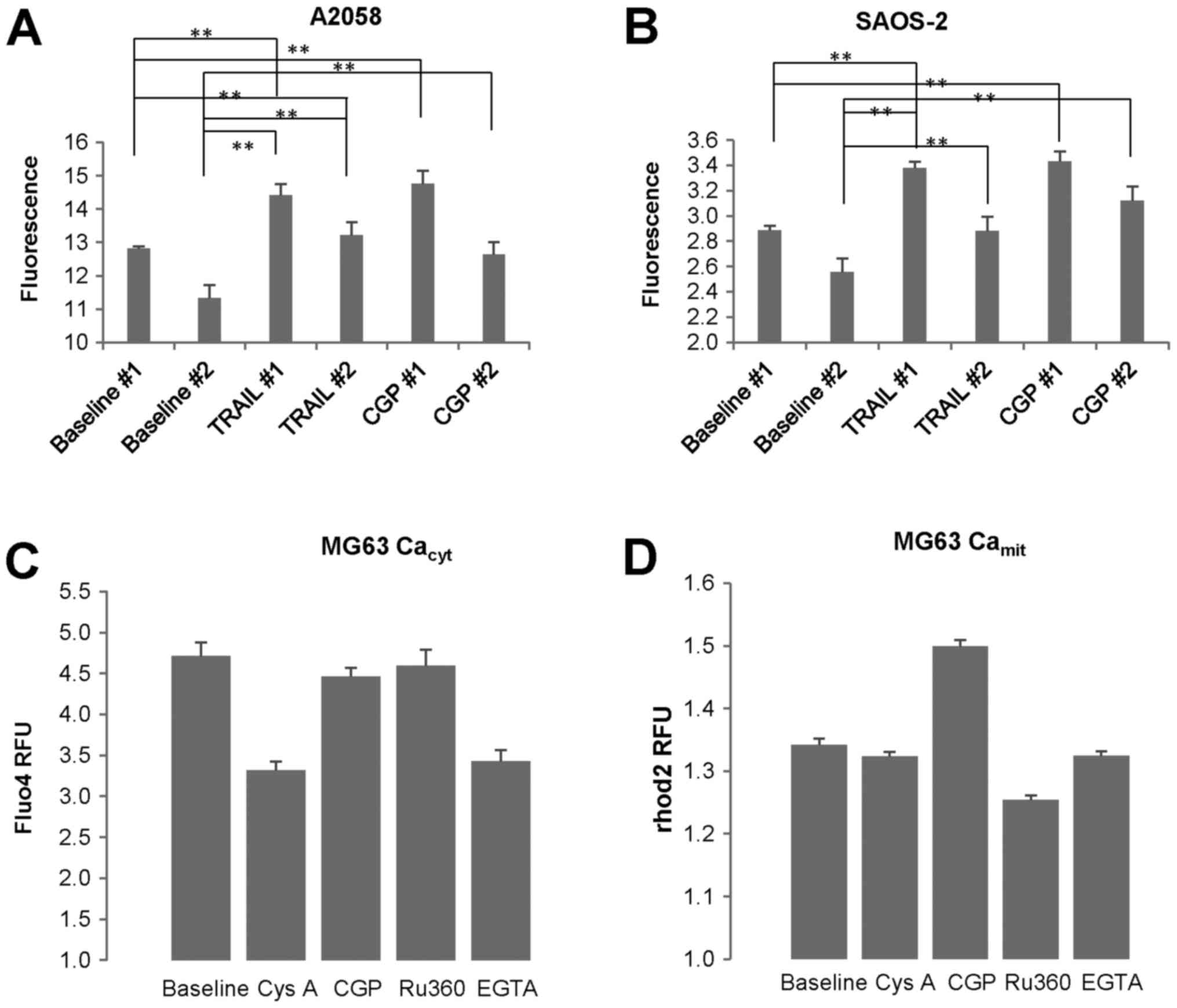
TRAIL-resistant cells. As mentioned above, in some experiments (Exp
#1) the basal [Ca2+]mit in SAOS-2 cells was
kept throughout the time monitored (10 min), while in other tests
(Exp #2) it was declined over time. In any case,
[Ca2+]mit became significantly higher in
response to CGP-37157 compared with the corresponding baseline
(Fig. 2A and B). We obtained
similar results in A2058 and all OS cell lines tested. CGP-37157
treatment increased [Ca2+]mit but not
[Ca2+]cyt (Fig.
2C and D). On the other hand, ruthenium 360 (Ru360) decreased
[Ca2+]mit, but not
[Ca2+]cyt, while EGTA and cyclosporine A
(CysA) reduced both [Ca2+]cyt and
[Ca2+]mit. These results indicate that
Ca2+ extrusion through NCLX plays a pivotal role in
regulating [Ca2+]mit in MM and OS cells.
CGP-37157 enhances TRAIL cytotoxicity in
a tumor-selective manner
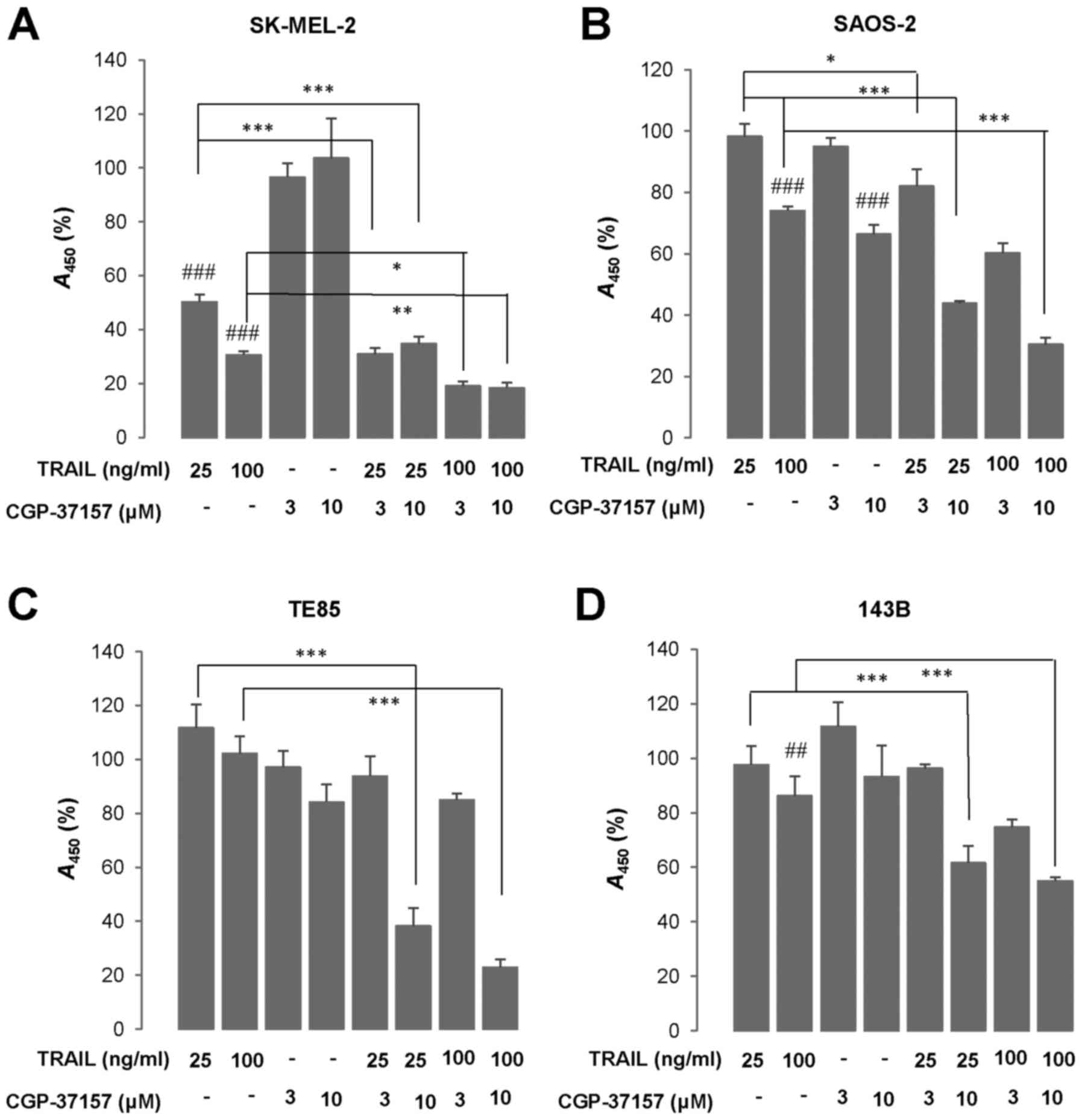
Next, we examined the effect of CGP-37157 on TRAIL
cytotoxicity. WST-8 assay revealed that treatment with TRAIL 25 and
100 ng/ml for 72 h led to a robust decrease in the viability of
SK-MEL-2 cells (50 and 70% reduction, respectively) (Fig. 3A). The three OS cell lines tested
(HOS, TE85, 143B) were highly resistant to TRAIL cytotoxicity while
SAOS-2 cells were moderately resistant. At 100 ng/ml TRAIL
decreased the viability of SAOS-2 cells moderately (maximum of 40%
reduction) (Fig. 3B). Whereas, the
TRAIL treatment led to only a modest decline (<15%) in their
viability or rather increased growth in TE85 and 143B cells
(Fig. 3C and D). CGP-37157
treatment ≤10 µM alone for 72 h minimally affected cell
growth. However, this compound significantly amplified TRAIL
cytotoxicity regardless of cancer cell types (Fig. 3A–D). We observed a smaller
amplification of TRAIL-induced cell death in moderately
TRAIL-sensitive MM and OS cell lines such as A375 and SAOS-2 cells
during the initial 24 h, and the pan-caspase-inhibitor Z-VAD-FMK
abrogated the effect completely (Fig.
3E and F). Moreover, flow cytometric analyses using a
caspase-3/7-specific substrate showed that CGP-37157 significantly
amplified TRAIL-induced caspase-3/7 activation (Fig. 3G). This effect became pronounced
over time. Simultaneous measurement using 7-AAD, a cell membrane
damage/death marker, revealed that the cell populations
corresponding to both early and late apoptotic cells were
increased, but the increases were blocked by Z-VAD-FMK only
partially. In contrast to MM and OS cells, TRAIL and CGP-37157
alone or in combination had the minimal effects on the growth of
WI-38 fibroblasts (Fig. 3H). These
results show that CGP-37157 enhances TRAIL cytotoxicity in a
tumor-selective manner.
OXOPHOS inhibitors increase and
cooperatively modulate [Ca2+]mit in MM and OS
cells
Classic OXOPHOS inhibitors such as antimycin A,
CCCP, and FCCP sensitize different tumor cell types including MM
and OS cells to TRAIL cytotoxicity (38–40).
These facts led us to investigate the possible impact of OXOPHOS
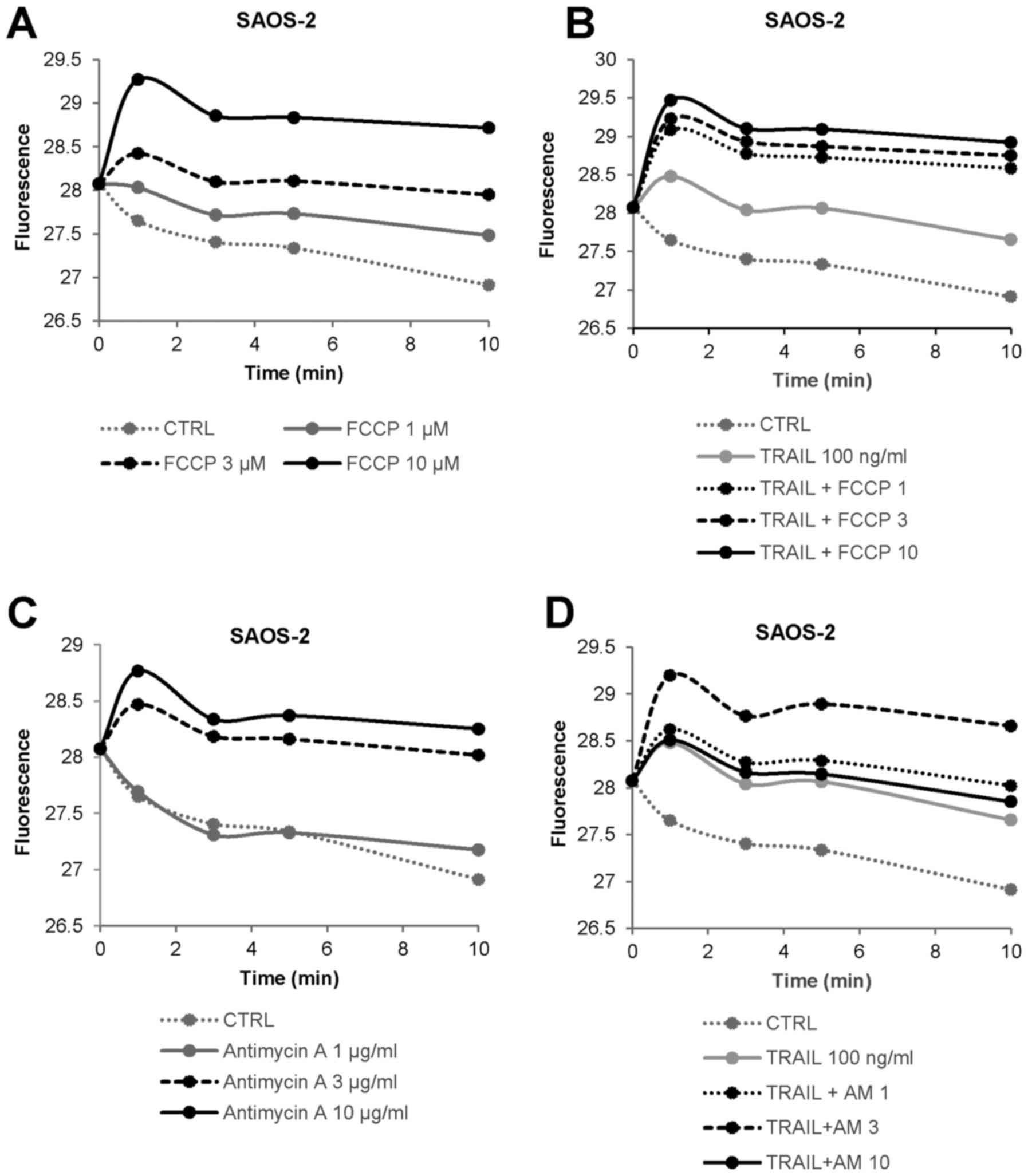
inhibitors on mitochondrial Ca2+ dynamics. FCCP at
concentrations ranging from 1 to 10 µM resulted in a rapid
and persistent increase in [Ca2+]mit in
SAOS-2 cells in a dose-dependent manner (Fig. 4A). The effect of 3 µM FCCP
was almost comparable to that of 100 ng/ml TRAIL (Fig. 4B). The combined use of TRAIL and
FCCP led to a greater extent of [Ca2+]mit
rise except for 10 µM FCCP compared with either agent alone.
Antimycin A also increased [Ca2+]mit in a
dose-dependent manner (Fig. 4C).
The agent at 3 µg/ml was equivalently potent to 100 ng/ml
TRAIL (Fig. 4D). Also, TRAIL +
antimycin A was more efficient than either agent alone in
increasing [Ca2+]mit except for 10
µg/ml of antimycin A, at which the combined use had a lesser
effect than antimycin A alone. We obtained similar results in A2058
and several OS cell lines (data not shown). These results indicate
that OXOPHOS inhibitors increase and cooperatively modulate
[Ca2+]mit in MM and OS cells.
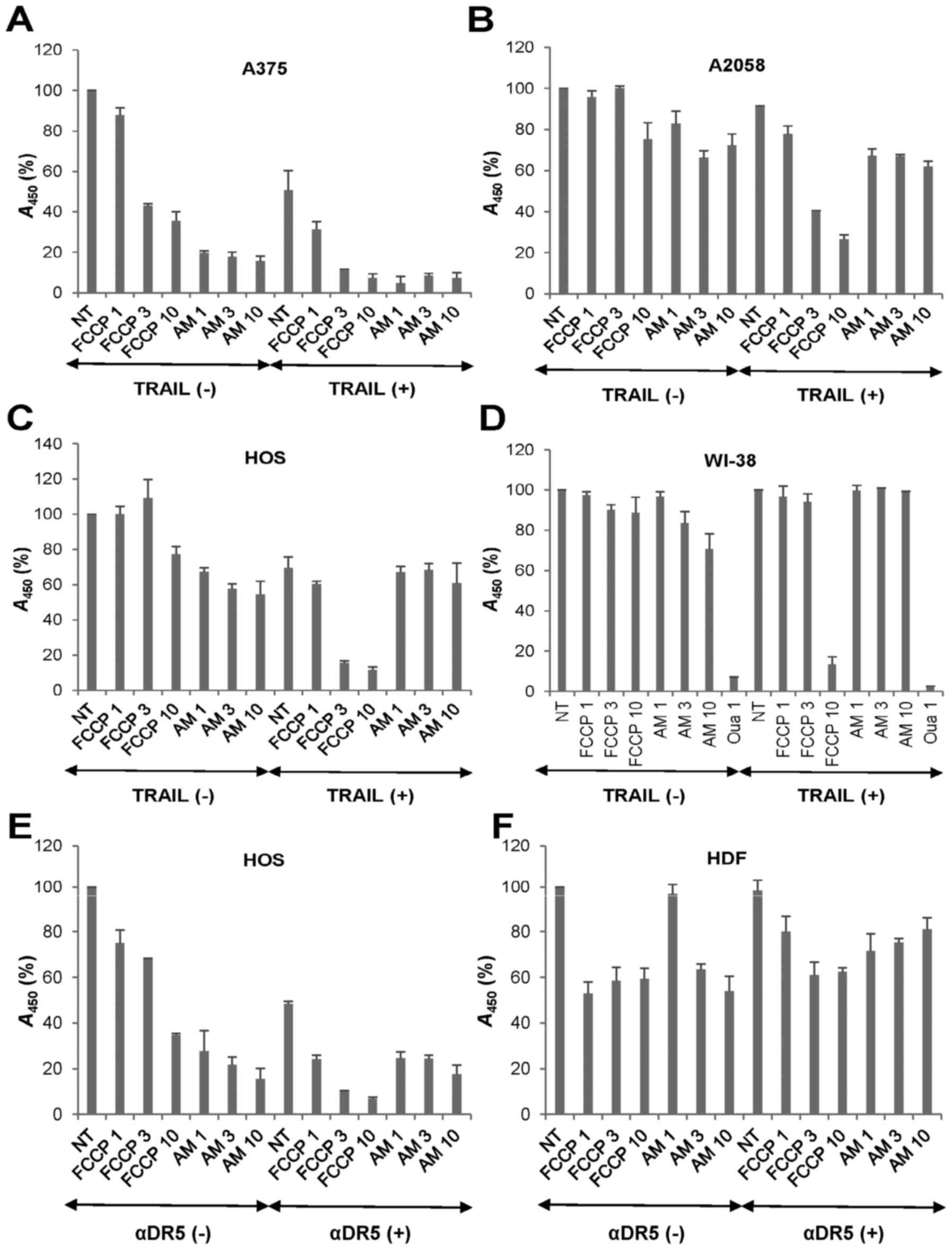
OXOPHOS inhibitors potentiate TRAIL
cytotoxicity in a tumor-selective manner
Then, we examined whether the OXOPHOS inhibitors
affected TRAIL cytotoxicity toward MM and OS cells. Treatment with
antimycin A (≥1 µg/ml) for 72 h decreased the viability of
A375 cells in a dose-dependent manner while FCCP (≥3 µM)
reduced it moderately (57%) (Fig.
5A). Moreover, the combined application of TRAIL and either
agent except for FCCP (1 µM) decreased the viability almost
entirely (>90% reduction). Highly TRAIL-resistant A2058 cells
were also more resistant to the cytotoxicity of all these OXOPHOS
inhibitors. As a result, antimycin A and FCCP at the maximal
concentrations caused only a modest decrease in their viability
(maximum of 30% decrease) (Fig.
5B). However, both antimycin A and FCCP enhanced TRAIL
cytotoxicity at the nontoxic concentrations. OS cell lines
including HOS cells were more resistant than MM cells to all these
OXOPHOS inhibitors. FCCP also sensitized the cells while antimycin
A had a slight sensitizing effect (Fig. 5C). To determine whether the effect
of OXOPHOS inhibitors is selective for tumor cells, we analyzed the
impact of TRAIL and OXOPHOS inhibitor alone or in combination on
the growth of non-transformed cells. FCCP and antimycin A ≤10
µM had the minimal effects on the growth of WI-38
fibroblasts (Fig. 5D). Treatment
with TRAIL + FCCP or TRAIL + antimycin A had a marginal effect on
their growth (maximum of 14% reduction at 10 µM FCCP). The
tolerance to TRAIL and OXOPHOS inhibitors alone or in combination
was specific since 1 µM ouabain heavily killed the cells.
Likewise TRAIL, the agonistic antibody against DR5 (αDR5) also
synergistically killed HOS cells with FCCP and antimycin A
(Fig. 5E). HDF was highly
resistant to αDR5 and either OXOPHOS inhibitor alone or in
combination (Fig. 5F). These
results indicate that OXOPHOS inhibitors potentiate TRAIL
cytotoxicity in a tumor-selective manner.
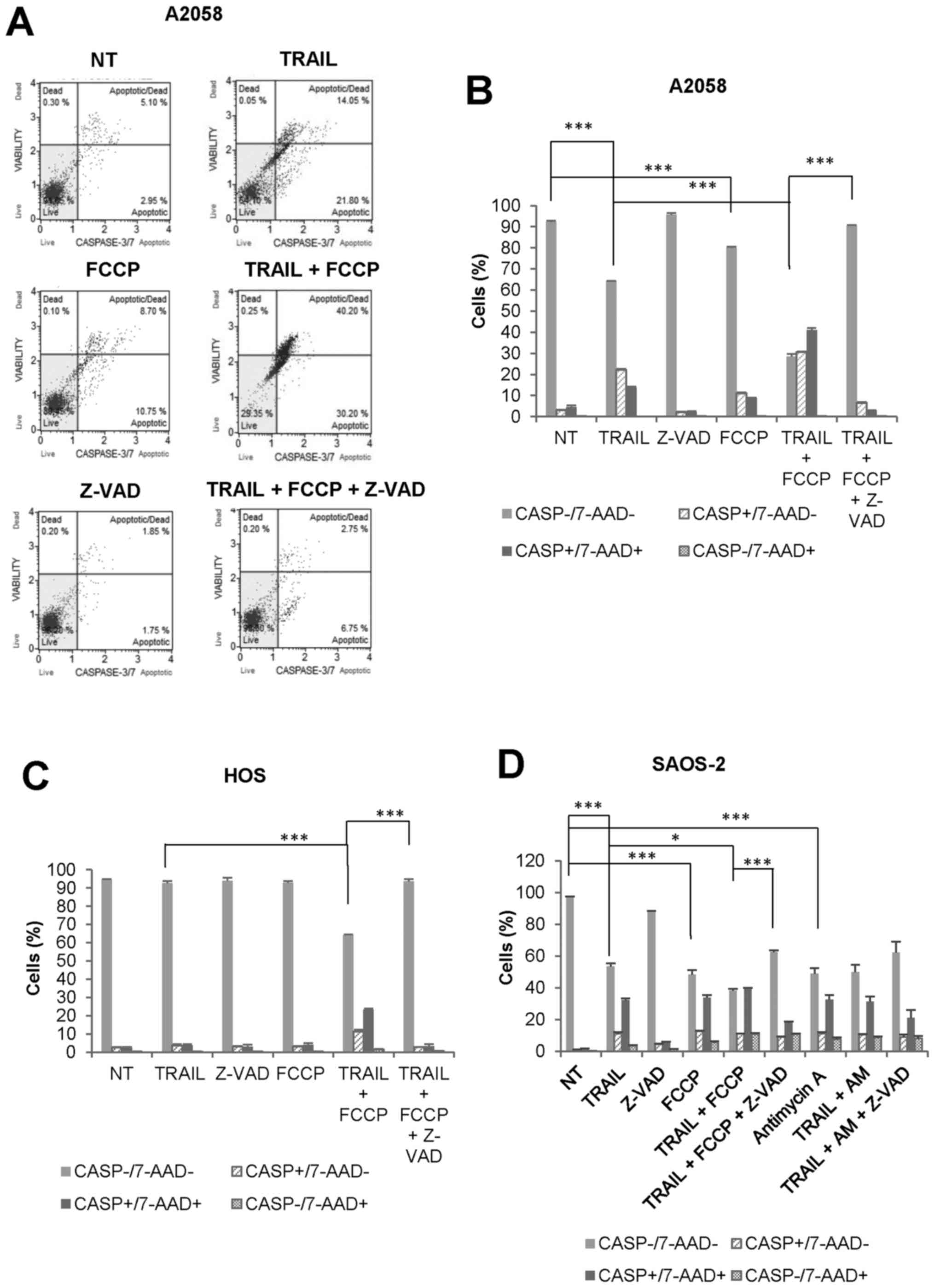
OXOPHOS inhibitors amplify both
caspase-dependent and caspase-independent cell death in MM and OS
cells at different time-points
To determine the cell death modality enhanced by
OXOPHOS inhibitors, we examined the effect of FCCP on caspase-3/7
activation and 7-AAD staining simultaneously. FCCP amplified
TRAIL-induced caspase-3/7 activation in A2058 cells during 24 h.
Both caspase-3/7-activated, 7-AAD-negative
(CASP+/7-AAD−) cells and
caspase-3/7-activated, 7-AAD-positive
(CASP+/7-AAD+) cells were increased, and
Z-VAD-FMK entirely inhibited the effects (Fig. 6A and B). Meanwhile,
caspase-3/7-inactivated, 7-AAD-positive
(CASP−/7-AAD+) cells were minimally increased
by TRAIL and FCCP alone or in combination. We obtained similar
results with HOS cells (Fig. 6C).
The cytotoxicity of TRAIL, as well as OXOPHOS inhibitors, became
more pronounced under prolonged incubation conditions (72 h).
Fig. 6D shows the representative
results obtained with SAOS-2 cells. TRAIL, FCCP, and antimycin A
reduced cell viability to a similar extent. Concomitantly,
CASP+/7-AAD+ cells were increased to
comparable levels. In addition, when TRAIL and FCCP applied in
combination, CASP−/7-AAD+ cells were
significantly increased compared with either agent alone
(10.95±0.35 for TRAIL + FCCP, 3.4±0.6 for TRAIL, 5.6±0.7 for FCCP,
p<0.01 vs TRAIL alone, P<0.05 vs FCCP alone, N=3). In
contrast, the effect of the combined application of TRAIL and
antimycin A was almost comparable to that of antimycin A alone.
Moreover, the cell death by TRAIL + either OXOPHOS inhibitor was
inhibited partially, but not completely by Z-VAD-FMK. Collectively,
these results indicate that OXOPHOS inhibitors amplify both
caspase-dependent and caspase-independent cell death at different
time-points.
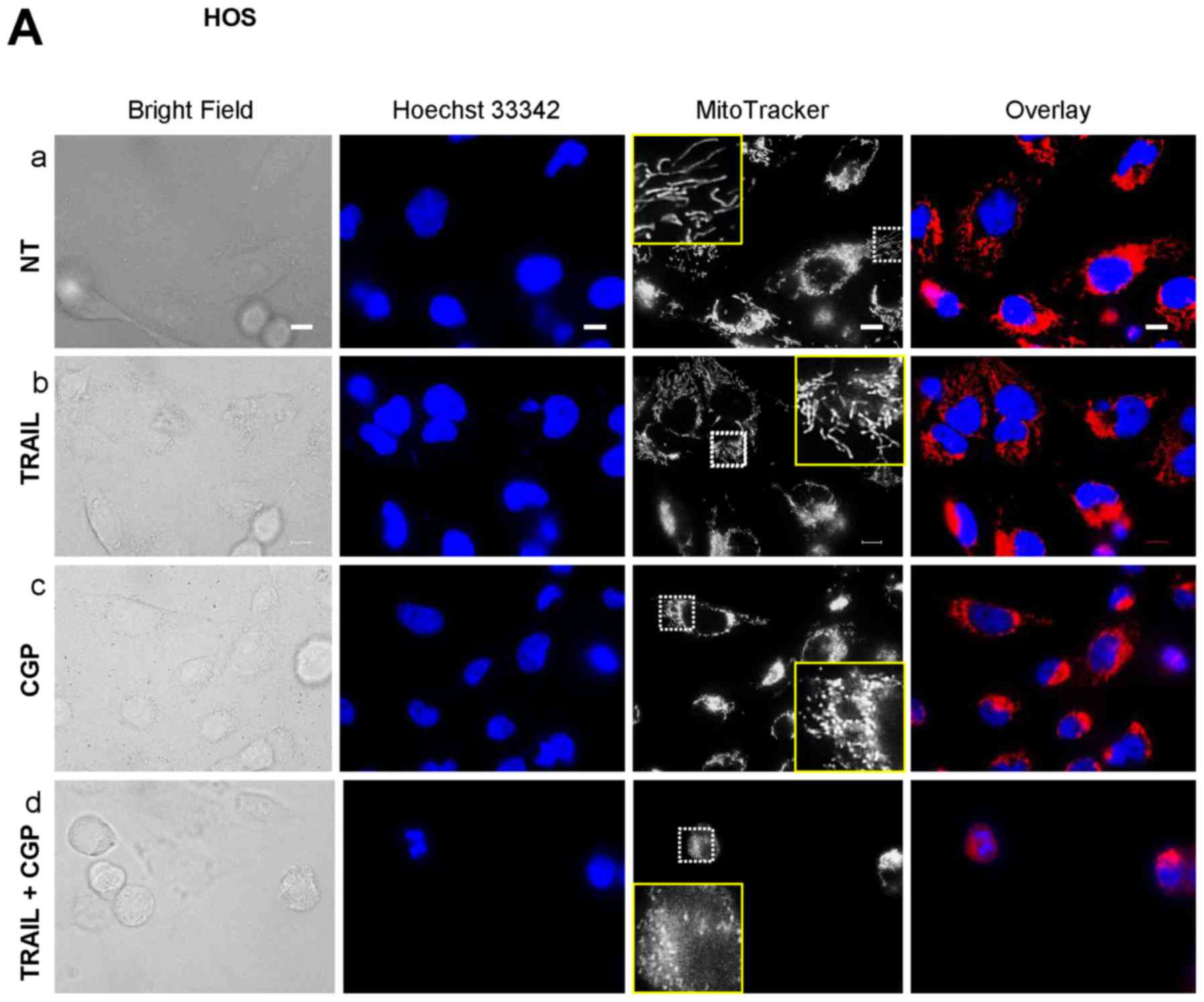
Mitochondrial Ca2+ overload
induces mitochondrial fragmentation and potentiates TRAIL-induced
mitochondrial network disruption
Previously, we reported that TRAIL modulates the
mitochondrial network dynamics in a tumor-selective manner and that
this effect is critical for TRAIL cytotoxicity (34). The facts led us to hypothesize that
the potentiation of TRAIL cytotoxicity might be related to
increased mitochondrial network abnormalities. To test this, we
analyzed the effect of the Ca2+-modulating agents on the
mitochondrial network using HOS cells as a model. Most healthy HOS
cells were highly adherent and possessed tubular mitochondria
around healthy nuclei (Fig. 7A-a).
Treatment with TRAIL for 24 h resulted in the minimal changes in
their morphology and a modest fragmentation of the mitochondria
(Fig. 7A-b) while CGP treatment
led to a substantial increase in round cells that possess punctate
mitochondria and damaged nuclei (Fig.
7A-c). When TRAIL and CGP-37157 were used in combination, the
mitochondria became more fragmented and clustered, and the nuclei
had smaller fragments (Fig. 7A-d).
Also, the red signals were overlapped with the blue signals,
thereby resulting in the appearance in the pink signals. FCCP and
antimycin A also led to a massive increase in round cells and
punctate mitochondria while the nuclei were damaged only modestly
(Fig. 7B-c and -d). Meanwhile,
when TRAIL and either agent were used together, most cells became
heavily damaged and detached from the coverslips. Accordingly, only
a small population of the damaged cells remained on the coverslips.
The remained cells possessed punctate, clustered mitochondria and
fragmented nuclei. Also, the overlapping of the red and the blue
signals became more pronounced (Fig.
7B-e and -f). These results indicate that mitochondrial
Ca2+ overload induces mitochondrial fragmentation and
potentiates TRAIL-induced mitochondrial network disruption.
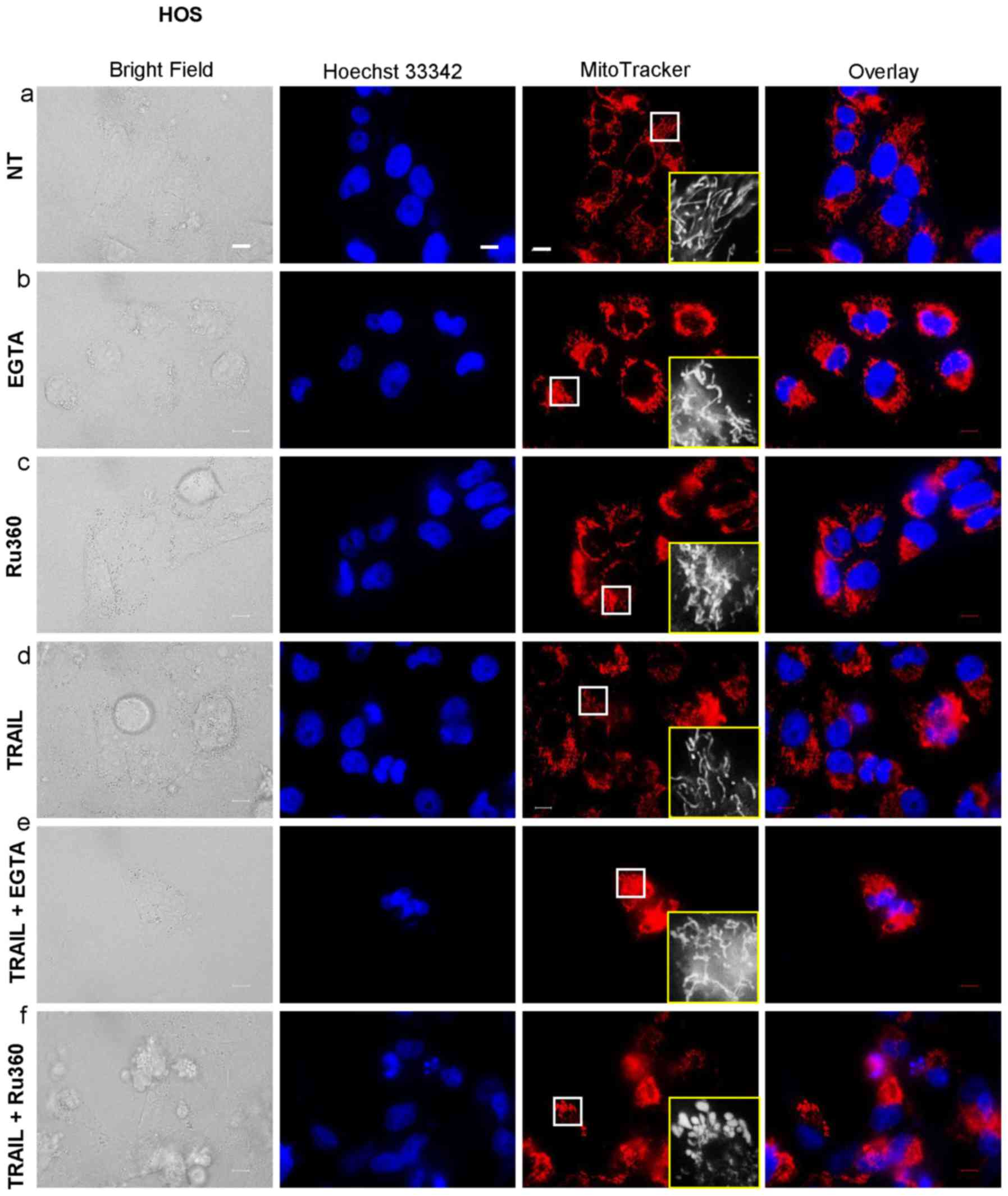
Mitochondrial Ca2+ removal
causes mitochondrial hyperfusion and potentiates TRAIL-induced
mitochondrial network disruption
The data presented above suggested that
mitochondrial Ca2+ overload promoted mitochondrial
fragmentation, suggesting that the Ca2+ regulates
mitochondrial fission positively. To further elucidate the
functional link between mitochondrial Ca2+ and the
mitochondrial dynamics, we examined the effects of EGTA and Ru360
on the mitochondrial network. Either EGTA or Ru360 alone led to a
robust mitochondrial hyperfusion, as indicated by the appearance in
elongated, highly interconnected mitochondria (Fig. 8-b and -c). Either agent alone had
the minimal effects on the cellular and nuclear morphology.
However, TRAIL + Ru360, but not TRAIL + EGTA, severely damaged the
cells and the mitochondria became punctate and clustered, and the
nuclei became fragmented (Fig.
8-f). Concomitantly, most red signals became overlapped with
the blue signals, thereby generating pink signals, as observed in
the case of the application of TRAIL + CGP or TRAIL + OXOPHOS
inhibitors. These results show that mitochondrial Ca2+
removal induces mitochondrial hyperfusion and eventually
potentiates TRAIL-induced mitochondrial network disruption.
Discussion
The present study demonstrated that TRAIL-resistant
MM and OS cells were tolerant to mitochondrial Ca2+
overload by the drug (Fig. 1).
Strikingly, the basal [Ca2+]mit spontaneously
declined over time in these cells, indicating the activation of a
certain mechanism for Ca2+ efflux from the mitochondrial
matrix. NCLX (35–37) and MPTPs (41,42)
participate in mitochondrial Ca2+ extrusion in normal
cells and some tumor cells. Therefore, it was possible that either
or both pathways contribute to the tolerance. Consistent with this
view, the present study demonstrates that NCLX plays a key role in
mitochondrial Ca2+ extrusion, thereby specifically
regulating [Ca2+]mit in MM and OS cells
(Fig. 2C and D). Accordingly,
CGP-37157 treatment led to a persistent mitochondrial
Ca2+ rise in all MM and OS cell lines tested. It is
well-known that mitochondrial Ca2+ has a dual function
depending on its magnitude and duration. Ca2+
accumulated in the mitochondrial matrix plays a critical role in
aerobic metabolism and cell survival while a persistent
Ca2+ overload is the primary cause of apoptosis
(17,19). The present study demonstrated that
CGP-37157 sensitized MM and OS cells to TRAIL cytotoxicity.
Z-VAD-FMK strongly blocked the effect, and CGP-37157 significantly
increased caspase-3/7 activation (Fig.
3). The results suggest that the persistent Ca2+
overload primarily promotes apoptosis. Our results are similar to
those reported by another group in prostate cancer cells (43,44).
The previous reports showed that CGP-37157 led to mitochondrial
Ca2+ overload and sensitized the cells to TRAIL-induced
apoptosis. Thus, NCLX seems to play a critical role in the
regulation of mitochondrial Ca2+ dynamics and TRAIL
sensitivity in cancer types from different origins. Moreover, we
found that OXOPHOS inhibitors such as FCCP and antimycin A also
caused mitochondrial Ca2+ overload (Fig. 4) and amplified TRAIL cytotoxicity
toward MM and OS cells (Fig. 5).
These two agents strongly sensitized all MM and OS cell lines
including those relatively tolerant to the effect of CGP-37157. As
expected, likewise CGP-37157, the OXOPHOS inhibitors significantly
enhanced TRAIL-induced caspase-3/7 activation, and Z-VAD-FMK
strongly blocked the effects (Fig.
6), supporting the view that apoptosis is the primary target in
the Ca2+-dependent TRAIL sensitization. Nevertheless,
our data suggested that mitochondrial Ca2+ overload
could also promote another caspase-independent cell death. It is
noteworthy that distinct cell death modalities seemed to be
encouraged by CGP-37157 and OXOPHOS inhibitors at different
time-points. At the early time (within 24 h of post-treatment),
apoptosis was mainly enhanced, as indicated by increased
caspase-3/7 activation and the complete blockade by the caspase
inhibitor (Figs. 3 and 6). Whereas, at the late time (72 h), the
additional nonapoptotic cell death was primarily amplified.
Interestingly, at that point OXOPHOS inhibitors alone caused a
substantial cell killing comparable to that induced by TRAIL, and
the simultaneous application of TRAIL had the minimal additional
cytotoxic effect. At present, the reason for such switching in the
cell death modality remains unclear. However, energy deprivation
under the prolonged cell cultivation might somewhat participate in
this switching. In support of this view, we noticed that similar
switching of cell death modalities from apoptosis to non-apoptotic
death occurred upon mitochondrial Ca2+ removal (24). Alternatively, autophagy caused by
energy depletion might modulate the cell death modality. Of note,
TRAIL induces autophagy in various cancer cell types, including MM
cells, and autophagy prevents apoptosis in these cells (45,46).
Alternatively, TRAIL might induce necroptosis, a non-apoptotic
programmed cell death, in the late stage since recent studies
revealed that TRAIL could induce both apoptosis and necroptosis
depending on the cellular caspase-8 activation and autophagy status
(9,47,48).
Mitochondrial Ca2+ overload has also been shown to cause
necrosis (17). In fact, we
observed a significant increase in the cell population with
necrotic cell death (CASP−/7-AAD+) upon
stimulation with TRAIL + the OXOPHOS inhibitors. However, the cell
population was still small (~11%), and our preliminary experiments
showed that necrostatin-1, a specific necroptosis inhibitor,
reduced the slow cell death only modestly. These results suggest
that necroptosis/necrosis plays a minor role in the slow cell
death, but further investigation is necessary to clarify the
role.
Another important finding in the present study is
that the Ca2+-dependent TRAIL sensitization functionally
links to the mitochondrial fission-fusion dynamics. Our results
demonstrated that all the Ca2+-modulating agents
markedly altered the mitochondrial dynamics. Specifically, agents
inducing mitochondrial Ca2+ overloads such as CGP-37157
and the OXOPHOS inhibitors led to mitochondrial fragmentation
(Fig. 7) while that causing
mitochondrial Ca2+ depletion such as EGTA and Ru360
evoked mitochondrial hyperfusion (Fig.
8). Moreover, regardless of their reciprocal actions on the
mitochondrial dynamics, the two types of agents commonly
exacerbated mitochondrial network collapse by TRAIL (Figs. 7 and 8). These findings strongly suggest that
an appropriate level of mitochondrial Ca2+ is essential
for maintaining the mitochondrial dynamics homeostasis. Our results
might provide insight into the controversial observations on the
role of mitochondrial fission in apoptosis. Some studies
demonstrate the requirement of mitochondrial fission machinery
including Drp1, Fis1, and Opa1 in pro-apoptotic events such as
cytochrome c release and apoptosis (28–30).
Whereas, other studies demonstrated that mitochondrial fission is
pro-survival and its inhibition promoted apoptosis (31–33).
It is noteworthy that at least in cancer cells such as prostate
(44) mitochondrial fragmentation
by itself is insufficient for inducing apoptosis. Our previous
study demonstrated that both an excess fragmentation of the
mitochondria and the subsequent clustering of the fragmented
mitochondria were essential for TRAIL-induced apoptosis in MM and
OS cells (34). The fragmentation
and clustering of the mitochondria occurred in a tumor-selective
manner and distinct from the usual Drp1-dependent mitochondrial
fission process because they were accelerated rather than reduced
by inhibition or knockdown of Drp1 (33). This observation strongly suggests
that Drp1-dependent reversible mitochondrial fission may prevent
the pro-apoptotic mitochondrial network abnormalities, thereby
serving as a pro-survival event. On the other hand, an excess
irreversible Drp1-independent mitochondrial fragmentation in
conjunction with abnormal clustering may be irreversibly committed
to mitochondrial dysfunction, thereby serving a pro-apoptotic
event. Consistent with this view, another group has reported that
mitochondrial aggregation preceded cytochrome c release and
apoptosis in arsenic trioxide-treated human glioblastoma cells
(49,50). Collectively, it is likely that an
appropriate level of mitochondrial Ca2+ is required for
the pro-survival reversible mitochondrial fission, thereby
preventing the pro-death mitochondrial network collapse. Further
studies to prove this scenario are underway in our laboratory.
In conclusion, we demonstrate in this report that
mitochondrial Ca2+ plays a vital role in maintaining the
mitochondrial dynamics and cell survival in MM and OS cells. Thus,
targeting mitochondrial Ca2+ homeostasis may serve as a
promising approach to overcome the TRAIL resistance of these
cancers without compromising the tumor-selectivity.
Acknowledgments
The authors thank Dr T. Ando for kindly providing
TE85 and 143B cells. We also appreciate Drs T. Tokunaga, T. Ito,
and A. Onoe for their technical assistance. This study was
supported in part by JSPS KAKENHI grant no. 15K09750 to Y.S.K.
References
|
1
|
Almasan A and Ashkenazi A: Apo2L/TRAIL:
Apoptosis signaling, biology, and potential for cancer therapy.
Cytokine Growth Factor Rev. 14:337–348. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herrero-Martín G, Høyer-Hansen M,
García-García C, Fumarola C, Farkas T, López-Rivas A and Jäättelä
M: TAK1 activates AMPK-dependent cytoprotective autophagy in
TRAIL-treated epithelial cells. EMBO J. 28:677–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He W, Wang Q, Xu J, Xu X, Padilla MT, Ren
G, Gou X and Lin Y: Attenuation of TNFSF10/TRAIL-induced apoptosis
by an autophagic survival pathway involving TRAF2- and
RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 8:1811–1821.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jouan-Lanhouet S, Arshad MI,
Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van
Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D,
Vandenabeele P, et al: TRAIL induces necroptosis involving
RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ.
19:2003–2014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sosna J, Philipp S, Fuchslocher Chico J,
Saggau C, Fritsch J, Föll A, Plenge J, Arenz C, Pinkert T, Kalthoff
H, et al: Differences and similarities in TRAIL- and tumor necrosis
factor-mediated necroptotic signaling in cancer cells. Mol Cell
Biol. 36:2626–2644. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol. 25(25): 4505–4506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
14
|
Guiho R, Biteau K, Heymann D and Redini F:
TRAIL-based therapy in pediatric bone tumors: How to overcome
resistance. Future Oncol. 11:535–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Miguel D, Lemke J, Anel A, Walczak H
and Martinez-Lostao L: Onto better TRAILs for cancer treatment.
Cell Death Differ. 23:733–747. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elustondo PA, Nichols M, Robertson GS and
Pavlov EV: Mitochondrial Ca(2+) uptake pathways. J Bioenerg
Biomembr. 49:113–119. 2017. View Article : Google Scholar
|
|
17
|
Bonora M, Wieckowski MR, Chinopoulos C,
Kepp O, Kroemer G, Galluzzi L and Pinton P: Molecular mechanisms of
cell death: Central implication of ATP synthase in mitochondrial
permeability transition. Oncogene. 34:1475–1486. 2015. View Article : Google Scholar
|
|
18
|
Izzo V, Bravo-San Pedro JM, Sica V,
Kroemer G and Galluzzi L: Mitochondrial permeability transition:
New findings and persisting uncertainties. Trends Cell Biol.
26:655–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Galluzzi L, Bravo-San Pedro JM, Kepp O and
Kroemer G: Regulated cell death and adaptive stress responses. Cell
Mol Life Sci. 73:2405–2410. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Calcium and mitochondria in the regulation of cell death. Biochem
Biophys Res Commun. 460:72–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Danese A, Patergnani S, Bonora M,
Wieckowski MR, Previati M, Giorgi C and Pinton P: Calcium regulates
cell death in cancer: Roles of the mitochondria and
mitochondria-associated membranes (MAMs). Biochim Biophys Acta.
1858:615–627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marchi S and Pinton P: Alterations of
calcium homeostasis in cancer cells. Curr Opin Pharmacol. 29:1–6.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Monteith GR, Prevarskaya N and
Roberts-Thomson SJ: The calcium-cancer signalling nexus. Nat Rev
Cancer. 17:367–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takata N, Ohshima Y, Suzuki-Karasaki M,
Yoshida Y, Tokuhashi Y and Suzuki-Karasaki Y: Mitochondrial
Ca2+ removal amplifies TRAIL cytotoxicity toward
apoptosis-resistant tumor cells via promotion of multiple cell
death modalities. Int J Oncol. 51:193–203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elgass K, Pakay J, Ryan MT and Palmer CS:
Recent advances into the understanding of mitochondrial fission.
Biochim Biophys Acta. 1833:150–161. 2013. View Article : Google Scholar
|
|
26
|
Chang CR and Blackstone C: Dynamic
regulation of mitochondrial fission through modification of the
dynamin-related protein Drp1. Ann NY Acad Sci. 1201:34–39. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Senft D and Ronai ZA: Regulators of
mitochondrial dynamics in cancer. Curr Opin Cell Biol. 39:43–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee YJ, Jeong SY, Karbowski M, Smith CL
and Youle RJ: Roles of the mammalian mitochondrial fission and
fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell.
15:5001–5011. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Estaquier J and Arnoult D: Inhibiting
Drp1-mediated mitochondrial fission selectively prevents the
release of cytochrome c during apoptosis. Cell Death Differ.
14:1086–1094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rehman J, Zhang HJ, Toth PT, Zhang Y,
Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C and Archer SL:
Inhibition of mitochondrial fission prevents cell cycle progression
in lung cancer. FASEB J. 26:2175–2186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Westrate LM, Sayfie AD, Burgenske DM and
MacKeigan JP: Persistent mitochondrial hyperfusion promotes G2/M
accumulation and caspase-dependent cell death. PLoS One.
9:e919112014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akita M, Suzuki-Karasaki M, Fujiwara K,
Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y and
Suzuki-Karasaki Y: Mitochondrial division inhibitor-1 induces
mitochondrial hyperfusion and sensitizes human cancer cells to
TRAIL-induced apoptosis. Int J Oncol. 45:1901–1912. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Suzuki-Karasaki Y, Fujiwara K, Saito K,
Suzuki-Karasaki M, Ochiai T and Soma M: Distinct effects of TRAIL
on the mitochondrial network in human cancer cells and normal
cells: Role of plasma membrane depolarization. Oncotarget.
6:21572–21588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nita II, Hershfinkel M, Kantor C, Rutter
GA, Lewis EC and Sekler I: Pancreatic β-cell Na+
channels control global Ca2+ signaling and oxidative
metabolism by inducing Na+ and Ca2+ responses
that are propagated into mitochondria. FASEB J. 28:3301–3312. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruiz A, Alberdi E and Matute C: CGP37157,
an inhibitor of the mitochondrial Na+/Ca2+ exchanger,
protects neurons from excitotoxicity by blocking voltage-gated
Ca2+ channels. Cell Death Dis. 5:e11562014. View Article : Google Scholar
|
|
37
|
Ben-Hail D, Palty R and Shoshan-Barmatz V:
Measurement of mitochondrial Ca2+ transport mediated by
three transport proteins: VDAC1, the Na+/Ca2+
exchanger, and the Ca2+ uniporter. Cold Spring Harb
Protoc. 2014:161–166. 2014.PubMed/NCBI
|
|
38
|
Izeradjene K, Douglas L, Tillman DM,
Delaney AB and Houghton JA: Reactive oxygen species regulate
caspase activation in tumor necrosis factor-related
apoptosis-inducing ligand-resistant human colon carcinoma cell
lines. Cancer Res. 65:7436–7445. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Inoue T and Suzuki-Karasaki Y:
Mitochondrial superoxide mediates mitochondrial and endoplasmic
reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells.
Free Radic Biol Med. 61:273–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suzuki-Karasaki M, Ochiai T and
Suzuki-Karasaki Y: Crosstalk between mitochondrial ROS and
depolarization in the potentiation of TRAIL-induced apoptosis in
human tumor cells. Int J Oncol. 44:616–628. 2014. View Article : Google Scholar
|
|
41
|
Bernardi P and von Stockum S: The
permeability transition pore as a Ca(2+) release channel: New
answers to an old question. Cell Calcium. 52:22–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gutiérrez-Aguilar M and Baines CP:
Structural mechanisms of cyclophilin D-dependent control of the
mitochondrial permeability transition pore. Biochim Biophys Acta.
1850:2041–2047. 2015. View Article : Google Scholar :
|
|
43
|
Kaddour-Djebbar I, Lakshmikanthan V,
Shirley RB, Ma Y, Lewis RW and Kumar MV: Therapeutic advantage of
combining calcium channel blockers and TRAIL in prostate cancer.
Mol Cancer Ther. 5:1958–1966. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kaddour-Djebbar I, Choudhary V, Brooks C,
Ghazaly T, Lakshmikanthan V, Dong Z and Kumar MV: Specific
mitochondrial calcium overload induces mitochondrial fission in
prostate cancer cells. Int J Oncol. 36:1437–1444. 2010.PubMed/NCBI
|
|
45
|
Han J, Hou W, Goldstein LA, Lu C, Stolz
DB, Yin XM and Rabinowich H: Involvement of protective autophagy in
TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem.
283:19665–19677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hou W, Han J, Lu C, Goldstein LA and
Rabinowich H: Enhancement of tumor-TRAIL susceptibility by
modulation of autophagy. Autophagy. 4:940–943. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Goodall ML, Fitzwalter BE, Zahedi S, Wu M,
Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD and
Thorburn A: The autophagy machinery controls cell death switching
between apoptosis and necroptosis. Dev Cell. 37:337–349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Haga N, Fujita N and Tsuruo T:
Mitochondrial aggregation precedes cytochrome c release from
mitochondria during apoptosis. Oncogene. 22:5579–5585. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Haga N, Fujita N and Tsuruo T: Involvement
of mitochondrial aggregation in arsenic trioxide
(As2O3)-induced apoptosis in human
glioblastoma cells. Cancer Sci. 96:825–833. 2005. View Article : Google Scholar : PubMed/NCBI
|