According to global cancer statistics for 2020, the
incidence and mortality rates of gastric cancer were the fifth and
fourth highest, respectively, among common cancers. In China, the
incidence and mortality rates of gastric cancer were both the third
highest among all cancers (1).
However, the occurrence of gastric cancer has not been studied in
depth, and its development is preceded by a series of progressive
changes, namely gastritis, atrophy, metaplasia, dysplasia and
carcinogenesis (2). Metaplasia is
defined as the replacement of a differentiated cell type by another
mature differentiated cell type that is not normally present in a
given tissue (3). In humans, two
types of precancerous metaplasia are associated with gastric
cancer, namely intestinal metaplasia (IM) and spasmolytic
polypeptide-expressing metaplasia (SPEM). SPEM, also known as
pseudopyloric metaplasia or mucinous metaplasia, is represented by
morphological features of the deep antral gland or Brunner's glands
and expresses trefoil factor 2 (TFF2) and mucin 6 (MUC6) (4,5).
IM is often considered a result of the transdifferentiation of
mucosa into an intestinal phenotype, which appears to be associated
with chronic inflammation (6) and
is characterized by the presence of mucin-containing goblet cells,
Paneth cells and absorptive cells and by the expression of TFF3 and
MUC2 (7,8). Another member of the trefoil protein
family, TFF1, is mainly secreted in gastric foveolar cells
(9) and has a crucial role in
gastrointestinal epithelium repair (10). The precise association between
SPEM and IM and their origins warrant further exploration in
large-scale clinical studies.
Gastric cancer is one of the most common cancers
worldwide, but it does not occur suddenly. Gastritis progresses to
malignant tumors after a series of developments and
differentiation. The most common pathological type of gastric
cancer is intestinal adenocarcinoma, which almost always occurs
after metaplasia and is considered to occur due to the loss of
acid-secreting parietal cells in the stomach (oxyntic atrophy)
(11). In 1999, Schmidt et
al (5) reported an abnormal
metaplasia cell lineage in the gastric fundic mucosa of mice
infected with Helicobacter felis, with morphological
features similar to those of Brunner's gland in the duodenum, which
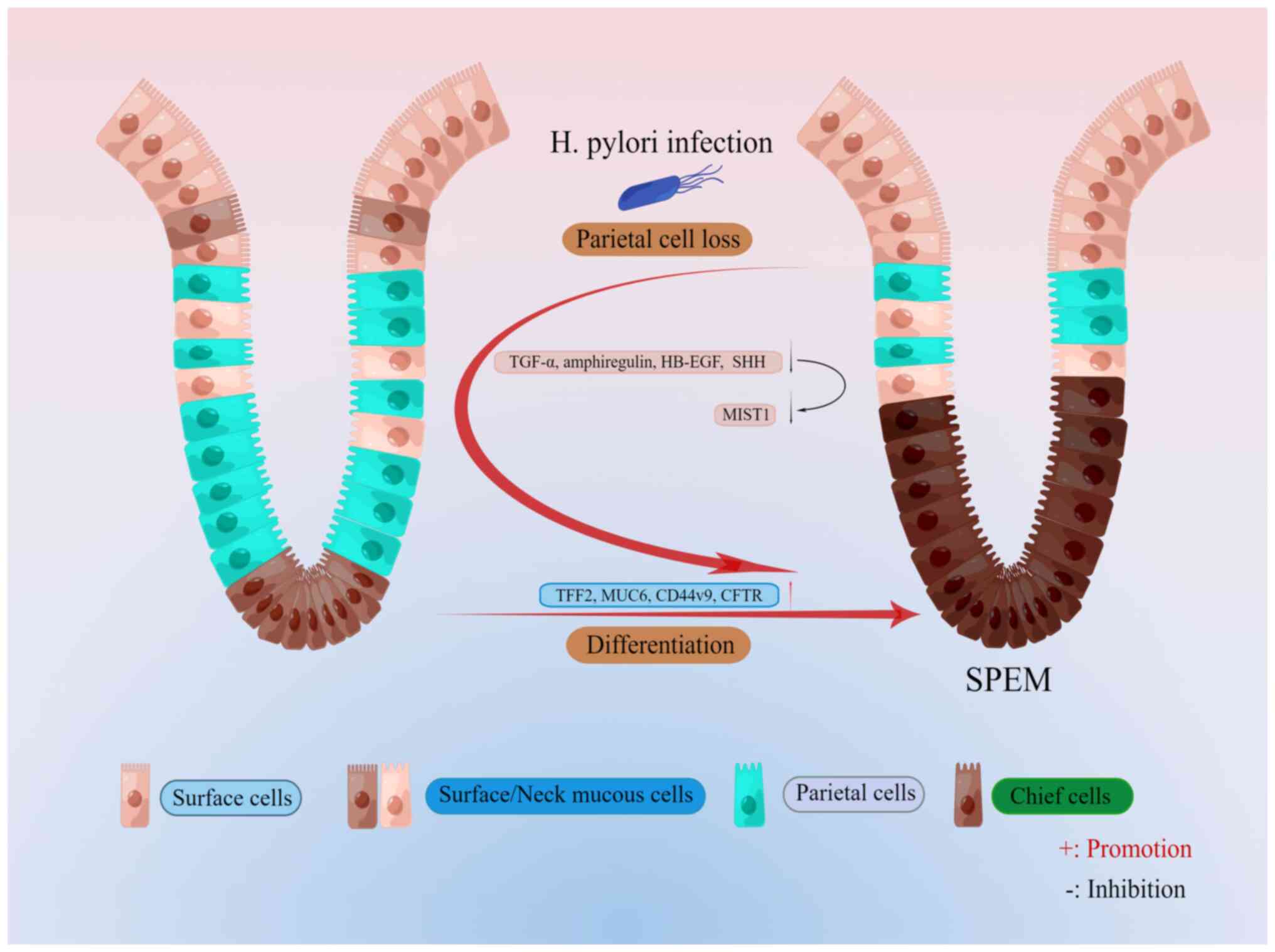
expressed TFF2, known as the spasmolytic polypeptide (Fig. 1). SPEM lineages have been further
characterized as diastase-periodic acid Schiff-positive lineages
expressing TFF2 and MUC6 and promoting GSII-agglutinin bonds,
similar to mucus-secreting cells of the deep antral glands
(12). Furthermore, this lineage
expresses several other anal genealogical markers, including CD44
variant 9 (CD44v9) and clusterin, where CD44v9 marks the SPEM in
the corpus and is involved in regeneration after gastric epithelial
cell injury (7,13,14). These studies have shown a high
degree of similarity between the spectra of SPEM and the deep
antral glands.
The stomach acts as an exocrine and endocrine organ
involved in the digestion of food. The gastric unit consists of the
glandular epithelium organized into repetitive tubular
invaginations (15). Each gastric
unit in the gastric body is divided into four regions: The apical
pit region, which consists of mucus-producing pit cells that
express MUC5AC and TFF1 (9); the
isthmus region (directly below the pit region), which contains
somatic stem cells expressing basic helix-loop-helix family member
a15 (Bhlha15; also known as MIST1, a transcription factor) or
transgenic markers driven by RUNX family transcription factor 1
enhancer elements (16,17); the neck region, which has neck
cells expressing MUC6 and TFF2 and the epitope for the lectin
Griffonia simplificolia (GS)II (9); and the bottom of the gastric somatic
unit, which is filled with chief cells expressing intrinsic factors
(18). In addition to the
widespread distribution of acid-producing parietal cells across all
four regions, it is essential to acknowledge that the emergence of
SPEM is intimately connected to the observed plasticity in chief
cells (18). Furthermore, there
is evidence indicating that the transformation into SPEM occurs not
only in the chief cell zone but also in the basal region (19,20). This highlights the intricate
mechanisms at play in the development of SPEM. The mature principal
cells in the isthmus region function as reserve stem cells in the
metaplastic process, capable of reprogramming into various cell
types. Regarding the topic of drug-induced SPEM, recent studies
suggest that these mature principal cells in the isthmus are the
primary contributors to the formation of SPEM cells, rather than
the isthmic progenitor cells (18) (Fig.
1). This discovery challenges prior beliefs and necessitates
further research for a comprehensive understanding of the
mechanisms involved. However, the recognition of this novel source
of SPEM cells marks a critical advancement in elucidating the
process of cellular transformation within the stomach.
The human stomach undergoes daily damage from
intrinsic or extrinsic sources, such as corrosion by gastric acid
and damage by food, and the stomach repairs the damaged area
through two mechanisms, namely superficial and glandular responses.
The superficial response of the stomach requires rapid adaptation
to prevent acid-induced epithelial integrity disruption. This
response relies on the secretion of local protective factors to
neutralize the corrosive effects of acid, regulation of mucosal
blood flow to limit the duration and extent of damage and
regeneration of the surface epithelium through recovery and
diffusion (21). In contrast to
the superficial response that occurs because of acid production,
the glandular response occurs when acid production is disrupted or
lost, and this response is characterized by a reduction in
acid-producing mural cells (atrophy). Furthermore, the glandular
response produces significant plasticity in gastric cells and this
plasticity increases the risk of cancer accumulation. However, the
main histological pattern observed in response to oxyntic atrophy
is gland reaggregation, which depletes mature mural and principal
cells but retains metaplastic cells (22). Thus, this glandular response can
also be considered the initiation of SPEM. The glandular response
can be transient or prolonged and the regulatory mechanisms are
unclear. The available literature indicates that SPEM is an
evolutionarily conserved mechanism in response to glandular injury,
but cellular signals and mechanisms regulating metaplastic
transitions in the stomach are still poorly understood.
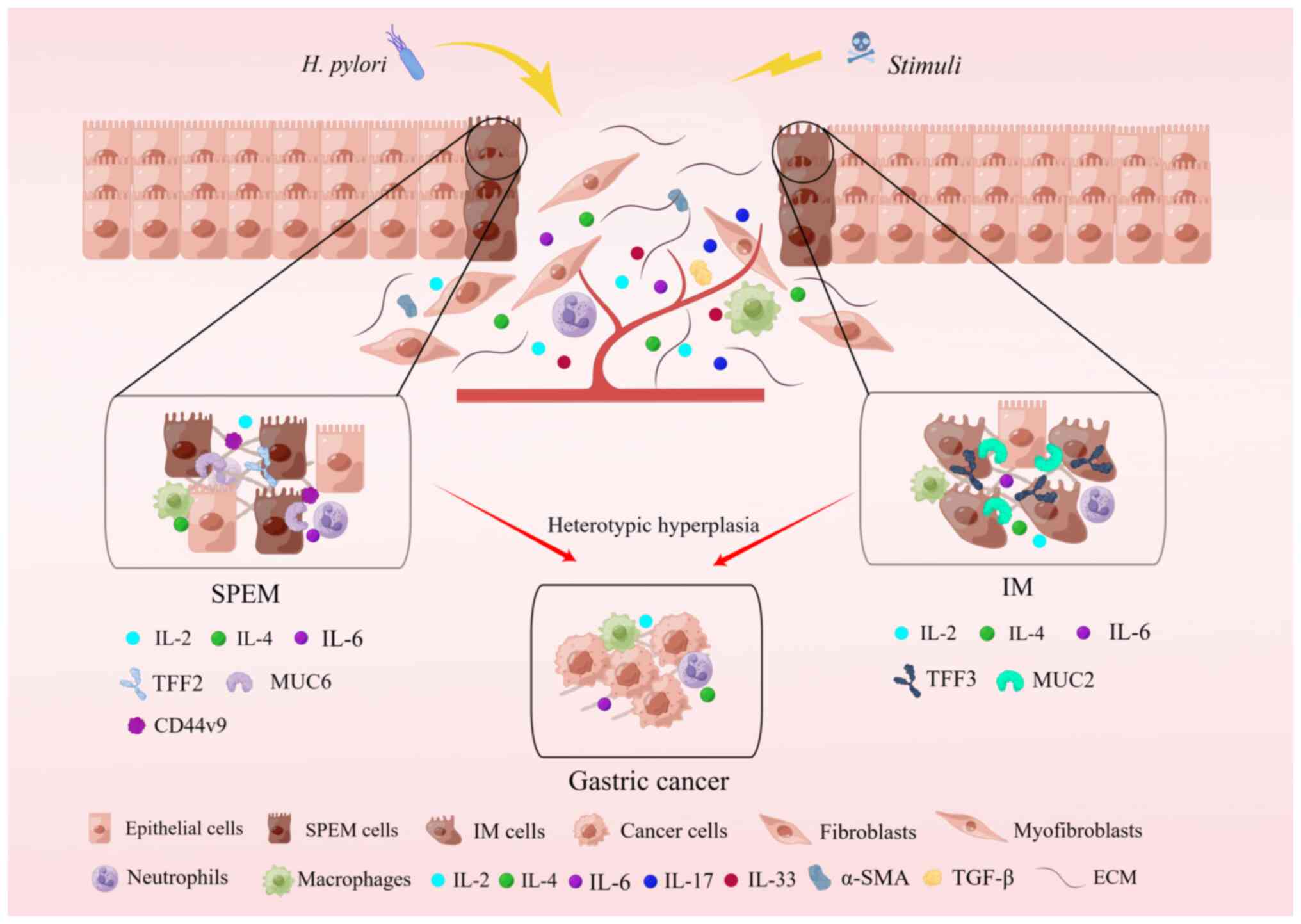
Oxyntic atrophy, accompanied by an inflammatory
response, can evolve into metaplasia, which manifests in two forms:
IM and SPEM, both linked with the progression to intestinal type
gastric cancer (5,29-32) (Fig.
2). Consequently, a pronounced inflammatory response and
oxyntic atrophy constitute the underlying factors for the altered
spectrum of gastric differentiation and the advancement toward
gastric cancer. Initially, IM was perceived as a precancerous
condition leading to intestinal-type cancers, characterized by IM
of cupped cells expressing specific intestinal markers such as MUC2
and TFF3 (7,8). Another form of precancerous
metaplasia, SPEM, was identified, exhibiting a mucinous metaplasia
profile. SPEM displays more distinct morphological characteristics
akin to deep anal gland cells or Brunner's glands, expressing
markers such as MUC6 and TFF2 (4,5).
Numerous studies have proposed that both SPEM and IM may represent
preneoplastic metaplastic conditions. Independent research
conducted in the US, Japan and Iceland has identified SPEM in
>90% of resected gastric cancers (5,32,33). Furthermore, SPEM is present in
almost all gastric cancer types and in studies on animal models
with chronic infection, transgenic and knockout genes, genetic
manipulation or acute oxyntic atrophy, the findings suggest that
SPEM is a crucial premalignant intermediate in the oncogenic
transformation of gastric cancer (34). However, the exact mechanism by
which SPEM promotes gastric carcinogenesis has remained elusive.
Furthermore, a controlled study demonstrated that patients with
gastric cancer infected with H. pylori had an increased
likelihood of developing precancerous SPEM in the stomach, but SPEM
regression could be achieved after the early eradication of H.
pylori (35-37). In addition, with the production
and regression of SPEM, some related tissue products, such as
miR-21, miR-155 and miR-223, showed an increase and decrease in
expression, respectively (36).
In humans, the origin of oxyntic atrophy and its
progression into gastric cancer are difficult to understand, but in
mouse experiments, these gaps can be addressed. Chronic H.
pylori infection is one of the major causes of gastric cancer
in humans, leading to oxyntic atrophy and marked inflammation.
Studies have shown that the loss of mural cells, downregulation of
the mature chief cell marker MIST1, and necroptosis of postmitotic
cells located at the base of the gastric gland are hallmarks of
SPEM (38). A mouse model of
chronic H. pylori infection can be used to observe H.
pylori infection in humans (39). After six months of Helicobacter
felis infection, mural cells were markedly lost with
inflammation, leading to the emergence of a proliferative SPEM
lineage, almost exclusively from transdifferentiated primary cells
(38), and SPEM develops into
atypical hyperplasia one year after infection onset without
phenotypic IM (40). It follows
that SPEM is a direct precursor of developmental abnormalities in
mice infected with H. felis. However, the presence of two
main causative factors, namely, oxyntic atrophy and marked
inflammation, make it difficult to elucidate the direct source of
SPEM from this model.
The role of mural cell loss and inflammation during
SPEM initiation may be distinguished using mural cell-specific
plasmid DMP-777-induced acute oxyntic atrophy in mice, L635-induced
acute inflammation in mice and chronic inflammation in mouse models
with H. felis infection (41,42). The plasmid DMP-777, which is a
cell-permeable inhibitor of neutrophilic leukocyte elastase, was
utilized to induce mural cell loss without eliciting an
inflammatory response. In mice, three days of DMP-777 treatment led
to oxyntic atrophy and 14 days of DMP-777 treatment induced SPEM as
a direct consequence of mural cell loss; however, even after one
year of DMP-777 treatment, SPEM did not progress to developmental
abnormalities (41). In addition,
DMP-777 caused acute mural cell loss along with increased serum
gastrin levels and altered mucosal dynamics (41,43). In another experiment, SPEM
developed more rapidly when gastrin- or amphipathic regulatory
protein-deficient mice were treated with DMP-777 (42,44). In comparison, L635, a proton
carrier analog of DMP-777 that lacks elastase inhibition, induced a
significant inflammatory response in mouse models and
phenotypically induced a spectrum of SPEM similar to that of H.
felis infection (38).
However, the progression of dysplasia cannot be analyzed through
the long-term administration of L635 at this time due to drug
availability problems. In studies of SPEM lineages, clusterin
expression was upregulated in all, with the most significant
increase in MUC5AC and MUC6 proteins (45), suggesting that clusterin
expression is a hallmark of SPEM regardless of the cause of SPEM
differentiation or the surrounding environment. In inflamed SPEM,
the upregulation of cystic fibrosis transmembrane conductance
regulator (CFTR) expression was observed, whereas CFTR expression
was not observed in normal gastric fundus and noninflamed SPEM
models (39). By contrast, in
human pathology, CFTR was not observed in SPEM, even when
accompanied by significant inflammation. However, IM featured
strong CFTR expression. Overall, the DMP-777 model revealed that
mural cell deficiency is sufficient to trigger SPEM, but
inflammation is essential for further SPEM differentiation and
development. Furthermore, the combination of the mouse model and
human pathology revealed an association between SPEM and
inflammation in mice and IM in humans (Fig. 2).
In addition, a study revealed that endogenous
glucocorticoids prevent gastric epithelial metaplastic transition
through the inhibition of spontaneous inflammation and are needed
to maintain gastric homeostasis (46). Glucocorticoids are steroid
hormones that inhibit proinflammatory stimuli, and adrenalectomy
results in the rapid onset of spontaneous gastritis, oxyntic
atrophy and metaplasia, expressing spasmolytic peptides (SPEM) in
mice. Regarding the process by which glucocorticoids prevent
metaplasia of the gastric epithelium, three days after
adrenalectomy, the proinflammatory genes monocyte chemokines C-C
motif chemokine ligand 2 and C-X3-C motif chemokine ligand 1 were
rapidly upregulated in the gastric body, and their expression in
the gastric body was inhibited by glucocorticoids (46). In addition, dysregulation of
monocytes and macrophages has been associated with the development
of several cancers; furthermore, glucocorticoids regulate monocyte
recruitment, regulate monocyte activity and prevent pathogenic
monocyte/lymphocyte activation (47,48). Numerous clinical studies have
demonstrated that patients with adrenocortical insufficiency
usually exhibit autoimmune and inflammatory gastritis, but
glucocorticoid replacement has been shown to suppress gastric
inflammation and metaplasia in mouse models with adrenocortical
insufficiency (49,50). However, a high dose of replacement
therapy may lead to epithelial cell damage and further induce SPEM
development (46,51,52). Thus, the disruption of
glucocorticoid signaling may lay a foundation for gastric cancer
development.
Oxyntic atrophy may lead to SPEM, but the cellular
origin of SPEM remains elusive. Of note, SPEM glands in human and
mouse stomachs are similar in many ways but need to be clearly
distinguished from IM because of their completely different
morphology and expression markers (39). Analyses of excised human gastric
body specimens showed that SPEM cells are present in the deeper
regions of the gland in areas containing complex glands, and an IM
lineage was observed in the luminal portion of the gland,
suggesting that IM may be formed by proliferative SPEM (53). The origin of gastric epithelial
metaplasia has been controversial in mouse models. Previously,
studies had proposed that significant proliferation occurs in the
isthmus of the stem cell zone, and therefore, metaplasia was
considered to originate from isthmus stem cells or progenitor cells
(53-55). Principal cells or a subset of
principal cells may transdifferentiate into SPEM cells (42,43,56). However, a recent study proposed
that mature chief cells are not needed for metaplasia development
(57). In that study, it was
proposed that the loss of chief cells is sufficient to cause
short-term SPEM-like lesions that originate from chief cell
precursors in the gastric neck region (57). Furthermore, certain phenotypic
chief cells expressing leucine-rich repeat-containing G-protein
coupled receptor 5 (Lgr5) were found at the lesser curvature of the
gastric body, which failed to promote short- and long-term
metaplasia, whereas isthmic stem and progenitor cells effectively
promoted long-term metaplasia. Studies have implied a major role of
chief cell precursors in the neck region in short-term gastric
regeneration and of isthmus stem or progenitor cells in long-term
metaplasia (16) (Fig. 1). It was further demonstrated that
SPEM occurs not through dedifferentiation of the principal cells
but through a compensatory response of neck cells to replace
eliminated principal cells (58).
Experiments with Lgr5-2A-CreERT, a variant of the enzyme Cre
derived from Escherichia coli, which has high recombination
efficiency and selectivity, mice revealed that Lgr5 principal cells
do not contribute to normal genealogical tracking or metaplasia
(59). Finally, it was concluded
that the expression of Lgr5 principal cells is irrelevant to any
SPEM development. Another study summarized the effect of microRNAs
(miRNAs) on the transdifferentiation of primary cells to SPEM
cells. Certain miRNAs were highly expressed in normal primary cells
but were downregulated in SPEM cells. Among these, miR-148a was
expressed >10-fold in primary cells, and in addition, miR-148a
deletion resulted in the upregulation of the early SPEM marker
CD44v9 and one of its target genes, DNA methyltransferase 1. These
findings suggest that miR-148a is an early regulator in the
reprogramming of chief cells during transdifferentiation into SPEM,
and it may be involved in chief cell maturation and maintenance, as
well as plasticity (60).
Of note, SPEM was found to always appear in the
intermediate zone and then spread along the border between the
gastric sinus and the basal zone, with structural distortion and
concave proliferation of the gland also being the most prominent in
the intermediate zone along the smaller curvature and expanding
continuously toward the larger curvature over a longer infection
period (64). This pattern is
similar to that observed in human saprophytic development (66). In addition, H. pylori
colonizes first at smaller curvatures in both mouse models and
humans. This localization of metaplasia initiation in rodents and
humans may be a consequence of the colonization pattern of H.
pylori infection.
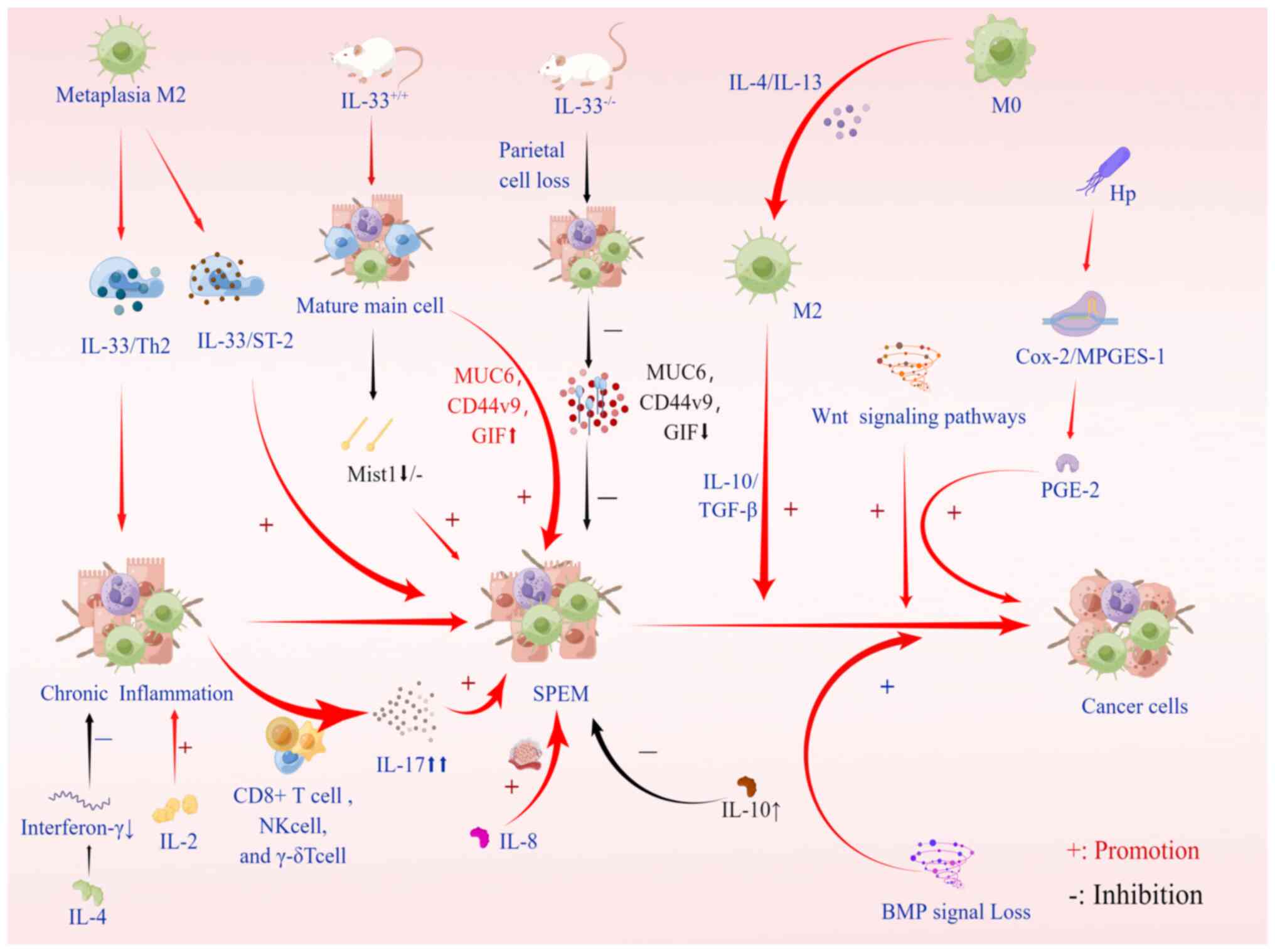
In the metagenesis process, inflammatory monocytes
can differentiate into M2 macrophage subsets under cytokine
stimulation, which can inhibit the immune response and promote
tissue remodeling through exposure to interleukin (IL)-4 and IL-13
and inactivated cytokines IL-10 and TGF-β (67). The M2 subpopulation of macrophages
is needed for metaplasia to develop into a more advanced SPEM
phenotype, and it penetrates the stomach following parietal cell
loss, thereby facilitating metaplasia (7). M2 macrophages are characterized as
anti-inflammatory, tumor-associated inflammatory cells driven by
type 2 T-helper (Th2) cytokines (68). Studies in mouse models and human
metaplasia have shown that M2 macrophages promote SPEM progression
in the context of inflammation and parietal cell atrophy (69). After RNA sequencing of macrophages
related to DMP-777 and L635 treatment in mice, IL-33 was found to
be significantly upregulated in late metaplasia M2 macrophages. The
IL-33/ST2 axis has a crucial role in the development of gastric
metaplasia and carcinogenesis. IL-33 is a key mediator in SPEM,
effectively stimulating epithelial cell proliferation and
metaplasia, and inducing the maintenance of Th2-driven chronic
inflammation, thereby increasing cancer risk (70,71). To demonstrate the necessity of
IL-33 and the IL-33 receptor (ST2) for SPEM induction, wild-type
mice were compared with IL-33-knockout (IL33KO) mice. It was
demonstrated that acute parietal cell loss in wild-type mice
results in the loss of MIST1, a mature chief cell transcription
factor, in chief cells located at the base of the gland (43). The results of the final experiment
showed that, compared with wild-type L635-treated mice,
L635-treated IL33KO mice expressed less MUC6 (GSII-lectin), CD44v9
and gastric intrinsic factor (GIF). Furthermore, in IL-33 receptor
(ST2) KO mice, SPEM deletion and proliferative SPEM decreased, and
the expression of MIST1 was reduced (7,70).
In addition, a study found that IL-13 has a central role in
supporting cytokines and producing immunomodulatory responses to
acute injury (70). Besides these
cytokines regulating the differentiation and development of
macrophages, deoxycholic acid has recently been documented to
promote SPEM development by altering macrophage secretion in mice
and modulating communication between macrophages and gastric
organs, thereby promoting SPEM development (72) (Fig.
3).
In addition to the two aforementioned ILs, numerous
other related ILs have been found to affect the occurrence and
development of SPEM, e.g., the proinflammatory cytokine IL-17A
secreted by CD4+ Th cells and other immune cells, such as CD8+ T
cells, natural killer cells and γ-δT cells (73-75). Related experiments have
demonstrated that parietal cells respond to IL-17A by undergoing
apoptosis, and excess IL-17A is produced during chronic
inflammation, thus promoting atrophy and metaplasia (76). In addition, IL-2 is a
proinflammatory cytokine that promotes the introduction and spread
of inflammatory immune responses, including H.
pylori-induced gastric inflammation (77). IL-4 is an anti-inflammatory
cytokine that inhibits gastric mucosa inflammation and atrophy by
reducing interferon-γ. IL-6, as a multifunctional cytokine,
regulates inflammatory mediators and endocrine responses and acts
as a messenger between the innate and adaptive systems in host
defense mechanisms. Its expression was found to increase in H.
pylori-associated gastritis and decrease after eradication
(78). Furthermore, IL-8 induces
cell proliferation, migration and angiogenesis (79). In the tamoxifen-induced SPEM
model, decreased IL-10 was found to be closely related to SPEM
occurrence. IL-10 may regulate gastric mucosa homeostasis, inhibit
mucogenesis development and have a potential therapeutic role in
the inhibition of SPEM development in early gastric cancer
(80). Further research into the
role of IL-10 in the epithelium may shed light on the mechanism by
which SPEM is activated in gastric tissue.
Bone morphogenetic protein (BMP) signaling in the
gastrointestinal tract is essential for the correct specification
of epithelial cell lineages and gastric endocrine cells (81). Some scientists have observed that
BMP signaling has a role in gastric tumorigenesis (82,83). However, in these experimental
models, the possible role of mesenchymal BMP signaling could not be
ruled out in their phenotypes. Further studies have speculated that
when BMP signaling is lost, induction of an inflammatory response
may lead to a series of drastic changes that increase sensitivity
to tumorigenesis (81). Studies
have suggested that BMP signaling may be involved in the terminal
differentiation of certain subsets of intestinal epithelial cells
(84). Furthermore, BMP signaling
may have a specific role in gastric epithelial cells. In the
stomach, BMP signaling negatively regulates endocrine cell
production. It also has a role in the control of gastric epithelial
cell proliferation, glandular morphogenesis and gastric cancer
development (81). In addition,
Wnt and prostaglandin E2 (PGE2) signaling pathways have been
studied. When activated, Wnt signaling leads to the development of
pretumor lesions. H. pylori infection induces the expression
of cyclooxygenase 2 and microsomal PGE synthase-1, which induces
PGE2 synthesis and leads to SPEM development. Experimental data
demonstrate that simultaneous activation of these two signaling
pathways leads to gastric tumor development through metaplastic
transitions (SPEM) and that the three signaling pathways are
correlated. Wnt signaling alone is not sufficient to promote tumor
development. Relevant experimental results indicated that tumor
formation in BMP-inhibited gastric mucosa also requires the
induction of the PGE2 pathway (85). In addition, huntingtin interacting
protein 1-related has a crucial role in SPEM formation in response
to gastric inflammation. It changes the metagenetic lineage of
gastric mucosa by affecting the hypertrophy and proliferation of
mucosal cells in the enzymatic lineage (86) (Fig.
3).
Gastric cancer may occur under specific conditions
after atrophy and metaplasia from H. pylori infection. Among
them, the role of inflammation is indispensable. It is now
suggested that the development of acute SPEM precedes oxyntic
atrophy and that the development of SPEM depends on gastric
infiltration of C-X3-C motif chemokine receptor 1 + monocytes
(46). These results provide
novel insight into the normal physiology of the stomach and the
mechanisms that regulate pathogenic gastric inflammation and
metaplastic transitions.
In mouse experiments, SPEM is produced after
drug-affected parietal cell loss but cannot be further cancerous
because of the lack of inflammatory stimulation. After a
significant inflammatory response, H. pylori-infected mice
further differentiate abnormally (87). Furthermore, a study of the
inflammatory component of mice revealed that the progression of
SPEM into developmental abnormalities requires a Th1-dominated
inflammatory response (88,89). In addition, a study showed that
bone marrow-derived cells (BMDCs) have a role in the progression of
metaplasia to developmental abnormalities, and during chronic H.
felis infection, BMDCs are recruited to the stomach, and these
recruited BMDCs appear to be transplanted into SPEM glands and
progress to deep cystic gastritis. Currently, it is uncertain
whether BMDC implantation into SPEM specifically targets H.
felis infection, and there is no strong evidence to support the
role of BMDCs in human metaplasia or carcinogenesis (40).
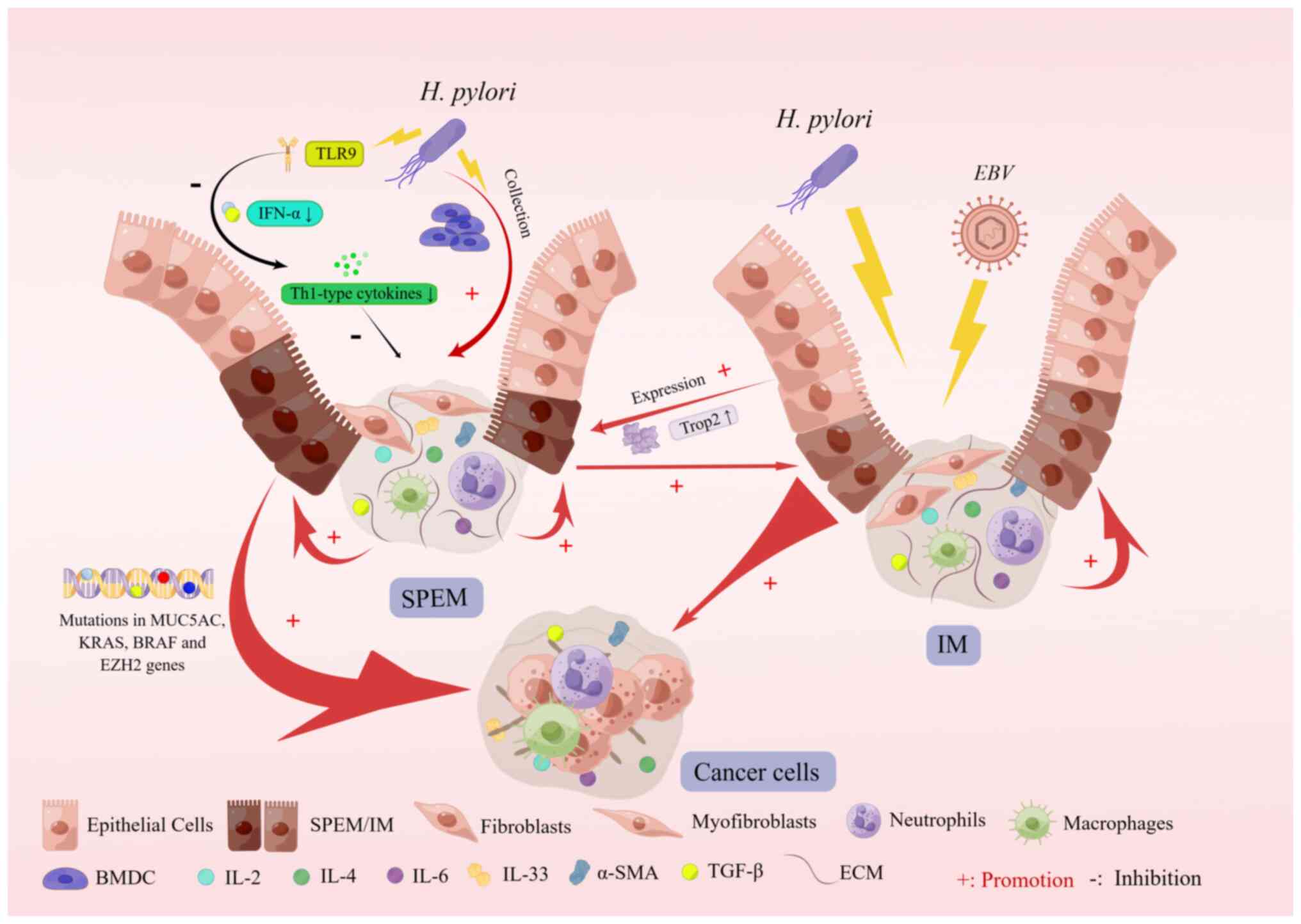
Recent experimental studies have shown that SPEM
development is accompanied by mutations in the MUC5AC, KRAS, BRAF
and enhancer of zeste 2 polycomb repressive complex 2 subunit genes
and that SPEM and gastric cancer are genomically similar (90). Furthermore, experimental studies
have shown that chronic H. pylori infection in both mice and
humans leads to the expression of Toll-like receptor 9 (TLR9),
which is associated with immunosuppression and an increased
incidence of gastric tumors in patients with TLR9 polymorphisms and
H. pylori infection (91-95). Studies of the target genes miR30a
and integrin α2 (ITGA2) revealed that miR30a expression was
downregulated in pretumor and early gastric cancer tissues but
maintained at a certain level in advanced gastric cancer tissues
and that miR30a had a significant tumor-suppressive effect, whereas
ITGA2 levels were significantly increased in gastric cancer tissues
(96). A pathological study of
metaplasia revealed that upregulation of trophoblast antigen 2
(TROP2) expression occurred during the transition from gastric
mucosal metaplasia to heteroplasia, and it promoted heteroplasia
(97) (Fig. 4).
In the proposed multistep pathway of Correa
carcinogenesis, mediated chronic gastritis progresses over the
years to atrophic gastritis, SPEM and IM, developmental
abnormalities and ultimately gastric cancer (98). Chronic H. pylori infection
is considered the underlying cause of IM and intestinal-type
gastric cancer (99). The
Epstein-Barr virus (EBV), the first virus associated with human
malignancy, is another powerful risk factor for gastric cancer
(100). Statistically,
EBV-associated gastric cancer (EBVaGC) accounts for ~9% of all
cases worldwide (101,102). In addition, H. pylori
infection is a crucial risk factor for gastric cancer, unlike
EBVaGC, suggesting that H. pylori and EBV are involved in
different carcinogenic pathways (103). The study was divided into two
clinical control studies-EBVaGC and EBV-negative gastric cancer
(EBVnGC)- and pathological sections of both types of patients were
subjected to laboratory tests, which ultimately revealed that both
EBVaGC and EBVnGC patients had a higher complication rate with SPEM
than with IM and that IM occurred more frequently in EBVnGC
patients, suggesting that the association between gastric cancer
and SPEM is stronger than IM. Furthermore, this study found that
early SPEM was more common in patients with EBVaGC, whereas late
SPEM was more common in patients with EBVnGC, and the different
distribution patterns of SPEM in these two groups of patients with
gastric cancer may be due to different pathogenic microbial
infections during gastric cancer (104).
In recent years, significant breakthroughs have been
achieved regarding the knowledge of gastric cancer development and
a deeper understanding was gained in this field. Studies of
metaplasia-associated genes have identified an independent
prognostic biomarker, calcium adhesion protein 17 (CDH17), which is
a structurally unique member of the calcium adhesion protein
superfamily and is a functional Ca2+-dependent
homologous cell adhesion molecule (105). A study reported that CDH17 was
markedly increased in SPEM and was expressed in 61-65% of human
gastric cancers. Although the relationship between CDH17 expression
and cancer stage or patient survival is inconclusive, it is now
considered an independent prognostic factor in patients with stage
I or lymph node-negative gastric cancer (106). SPEM has a crucial role in the
entire process of gastric cancer progression. The appearance of
SPEM has been found to precede intestinal epithelial hyperplasia in
numerous studies; therefore, the detection of early SPEM has a
clear role in gastric cancer diagnosis. Therefore, the
identification of markers of epithelial hyperplasia and hyperplasia
progression to atypical hyperplasia is necessary for the
development of effective screening methods that can identify
preproliferation. Several researchers have started investigating
the relationship between biomarkers and cancer. For instance, a
previously identified SPEM-specific marker, human epididymis
protein 4 (HE4), was initially used as a serum marker for ovarian
cancer, but as research progressed, is was found that HE4 is not
present in normal gastric sinuses but can significantly increase in
metaplasia and carcinoma, and this study identified secreted whey
acidic protein structural domain protein HE4, which may be used as
a putative biomarker (43,107).
Furthermore, HE4 was expressed in all SPEM and IM samples. Two
other early marker proteins of SPEM and IM have been recently
identified, lactotransferrin (LTF) and deleted in malignant brain
tumor 1 (DMBT1), both of which are associated with the inflammatory
response and cell differentiation. The expression patterns of LTF
and other SPEM markers, as well as DMBT1 support the notion that
human SPEM evolves into IM (108). Multiple types of BMDCs are
involved during SPEM development and the ability to track these
cell types in the preneoplastic state expands the options for more
effective screening of subjects susceptible to the eventual
occurrence of gastric cancer and the development of atrophic
gastritis when prophylactic treatment options, including mTOR
antagonists, are available (109). Gastrokine 3 (GKN3) mRNA can
accurately assess SPEM in the analysis of mouse and human chronic
inflammatory gastric tissues, mainly because it is absent in normal
gastric tissues. Furthermore, GKN3 mRNA and GKN3-positive cells
were detected in the gastric body during SPEM (110). In addition, human SPEM cells in
TROP2-labeled incomplete IM glands at the base strongly express
aquaporin 5 (AQP5) but not in intact IM glands, and AQP5-expressing
SPEM cells are present in pyloric metaplasia and TROP2-positive
incomplete IM, which may be an essential component of metaplasia
and can predict a higher risk of gastric cancer development
(111). Therefore, GKN3 mRNA and
AQP5 may be used as specific markers for SPEM diagnosis.
In summary, the present study described the role of
various cytokines and expression products in SPEM formation, and
with the discovery of specific markers, the understanding of the
origin of SPEM and cancer progression will improve, which may aid
in the easy and accurate diagnosis of early developmental
abnormalities. These results provide research directions on SPEM
pathogenesis and new opportunities for future diagnosis and
treatment.
Not applicable.
YC, DY, ZL and FN conceived and designed the study.
YC, DY and ZL performed the literature search. YC and DY drafted
the manuscript. YC, DY and FN designed and drew the figures. YC
critically revised the manuscript. All the authors were involved in
revising the paper critically. All authors have read and agreed to
the published version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The figures were generated with Figdraw.
The present study was funded by the National Natural Science
Foundation of China (grant nos. 81802792 and 81973518), Project of
Yangzhou University Medical Innovation and Transformation Special
Fund New Medical Cross Innovation Team (grant no. AHYZUCXTD
202108), Postgraduate Research & Practice Innovation Program of
Jiangsu Province (grant no. SJCX22_1822), Post-doctoral Science
Foundation of Jiangsu Province (grant no. 2020Z409) and Science and
Technology Projects for Social Development of Yangzhou City (grant
no. YZ2022106).
|
1
|
Cao W, Chen HD, Yu YW, Li N and Chen WQ:
Changing profiles of cancer burden worldwide and in China: A
secondary analysis of the global cancer statistics 2020. Chin Med J
(Engl). 134:783–791. 2021.
|
|
2
|
Kinoshita H, Hayakawa Y and Koike K:
Metaplasia in the stomach-precursor of gastric cancer? Int J Mol
Sci. 18:20632017.
|
|
3
|
Giroux V and Rustgi AK: Metaplasia: Tissue
injury adaptation and a precursor to the dysplasia-cancer sequence.
Nat Rev Cancer. 17:594–604. 2017.
|
|
4
|
Weis VG and Goldenring JR: Current
understanding of SPEM and its standing in the preneoplastic
process. Gastric Cancer. 12:189–197. 2009.
|
|
5
|
Schmidt PH, Lee JR, Joshi V, Playford RJ,
Poulsom R, Wright NA and Goldenring JR: Identification of a
metaplastic cell lineage associated with human gastric
adenocarcinoma. Lab Invest. 79:639–646. 1999.
|
|
6
|
Barros R, Freund JN, David L and Almeida
R: Gastric intestinal metaplasia revisited: Function and regulation
of CDX2. Trends Mol Med. 18:555–563. 2012.
|
|
7
|
Petersen CP, Weis VG, Nam KT, Sousa JF,
Fingleton B and Goldenring JR: Macrophages promote progression of
spasmolytic polypeptide-expressing metaplasia after acute loss of
parietal cells. Gastroenterology. 146:1727–1738 e8. 2014.
|
|
8
|
Nam KT, Lee HJ, Mok H, Romero-Gallo J,
Crowe JE Jr, Peek RM Jr and Goldenring JR: Amphiregulin-deficient
mice develop spasmolytic polypeptide expressing metaplasia and
intestinal metaplasia. Gastroenterology. 136:1288–1296. 2009.
|
|
9
|
Lefebvre O, Wolf C, Kédinger M, Chenard
MP, Tomasetto C, Chambon P and Rio MC: The mouse one P-domain (pS2)
and two P-domain (mSP) genes exhibit distinct patterns of
expression. J Cell Biol. 122:191–198. 1993.
|
|
10
|
Playford RJ, Marchbank T, Goodlad RA,
Chinery RA, Poulsom R and Hanby AM: Transgenic mice that
overexpress the human trefoil peptide pS2 have an increased
resistance to intestinal damage. Proc Natl Acad Sci USA.
93:2137–2142. 1996.
|
|
11
|
Mills JC and Goldenring JR: Metaplasia in
the stomach arises from gastric chief cells. Cell Mol Gastroenterol
Hepatol. 4:85–88. 2017.
|
|
12
|
Lennerz JK, Kim SH, Oates EL, Huh WJ,
Doherty JM, Tian X, Bredemeyer AJ, Goldenring JR, Lauwers GY, Shin
YK and Mills JC: The transcription factor MIST1 is a novel human
gastric chief cell marker whose expression is lost in metaplasia,
dysplasia, and carcinoma. Am J Pathol. 177:1514–1533. 2010.
|
|
13
|
Wada T, Ishimoto T, Seishima R,
Tsuchihashi K, Yoshikawa M, Oshima H, Oshima M, Masuko T, Wright
NA, Furuhashi S, et al: Functional role of CD44v-xCT system in the
development of spasmolytic polypeptide-expressing metaplasia.
Cancer Sci. 104:1323–1329. 2013.
|
|
14
|
Bertaux-Skeirik N, Wunderlich M, Teal E,
Chakrabarti J, Biesiada J, Mahe M, Sundaram N, Gabre J, Hawkins J,
Jian G, et al: CD44 variant isoform 9 emerges in response to injury
and contributes to the regeneration of the gastric epithelium. J
Pathol. 242:463–475. 2017.
|
|
15
|
Karam SM and Leblond CP: Identifying and
counting epithelial cell types in the 'corpus' of the mouse
stomach. Anat Rec. 232:231–246. 1992.
|
|
16
|
Hayakawa Y, Ariyama H, Stancikova J,
Sakitani K, Asfaha S, Renz BW, Dubeykovskaya ZA, Shibata W, Wang H,
Westphalen CB, et al: Mist1 expressing gastric stem cells maintain
the normal and neoplastic gastric epithelium and are supported by a
perivascular stem cell niche. Cancer Cell. 28:800–814. 2015.
|
|
17
|
Matsuo J, Kimura S, Yamamura A, Koh CP,
Hossain MZ, Heng DL, Kohu K, Voon DC, Hiai H, Unno M, et al:
Identification of stem cells in the epithelium of the stomach
corpus and antrum of mice. Gastroenterology. 152:218–231 e14.
2017.
|
|
18
|
Caldwell B, Meyer AR, Weis JA, Engevik AC
and Choi E: Chief cell plasticity is the origin of metaplasia
following acute injury in the stomach mucosa. Gut. 71:1068–1077.
2022.
|
|
19
|
Radyk MD, Burclaff J, Willet SG and Mills
JC: Metaplastic cells in the stomach arise, independently of stem
cells, via dedifferentiation or transdifferentiation of chief
cells. Gastroenterology. 154:839–843 e2. 2018.
|
|
20
|
Hayakawa Y, Fox YG and Wang TC: Isthmus
stem cells are the origins of metaplasia in the gastric corpus.
Cell Mol Gastroenterol Hepatol. 4:89–94. 2017.
|
|
21
|
Sáenz JB and Mills JC: Acid and the basis
for cellular plasticity and reprogramming in gastric repair and
cancer. Nat Rev Gastroenterol Hepatol. 15:257–273. 2018.
|
|
22
|
Goldenring JR and Nam KT: Oxyntic atrophy,
metaplasia, and gastric cancer. Prog Mol Biol Transl Sci.
96:117–131. 2010.
|
|
23
|
Blaser MJ and Parsonnet J: Parasitism by
the 'slow' bacterium Helicobacter pylori leads to altered gastric
homeostasis and neoplasia. J Clin Invest. 94:4–8. 1994.
|
|
24
|
Jain RN, Brunkan CS, Chew CS and Samuelson
LC: Gene expression profiling of gastrin target genes in parietal
cells. Physiol Genomics. 24:124–132. 2006.
|
|
25
|
Beauchamp RD, Barnard JA, McCutchen CM,
Cherner JA and Coffey RJ Jr: Localization of transforming growth
factor alpha and its receptor in gastric mucosal cells.
Implications for a regulatory role in acid secretion and mucosal
renewal. J Clin Invest. 84:1017–1023. 1989.
|
|
26
|
Murayama Y, Miyagawa J, Higashiyama S,
Kondo S, Yabu M, Isozaki K, Kayanoki Y, Kanayama S, Shinomura Y,
Taniguchi N, et al: Localization of heparin-binding epidermal
growth factor-like growth factor in human gastric mucosa.
Gastroenterology. 109:1051–1059. 1995.
|
|
27
|
Abe S, Sasano H, Katoh K, Ohara S, Arikawa
T, Noguchi T, Asaki S, Yasui W, Tahara E, Nagura H and Toyota T:
Immunohistochemical studies on EGF family growth factors in normal
and ulcerated human gastric mucosa. Dig Dis Sci. 42:1199–1209.
1997.
|
|
28
|
El-Zimaity HM, Ota H, Graham DY, Akamatsu
T and Katsuyama T: Patterns of gastric atrophy in intestinal type
gastric carcinoma. Cancer. 94:1428–1436. 2002.
|
|
29
|
Filipe MI, Muñoz N, Matko I, Kato I,
Pompe-Kirn V, Jutersek A, Teuchmann S, Benz M and Prijon T:
Intestinal metaplasia types and the risk of gastric cancer: A
cohort study in Slovenia. Int J Cancer. 57:324–329. 1994.
|
|
30
|
Xia HH, Kalantar JS, Talley NJ, Wyatt JM,
Adams S, Chueng K and Mitchell HM: Antral-type mucosa in the
gastric incisura, body, and fundus (antralization): A link between
Helicobacter pylori infection and intestinal metaplasia? Am J
Gastroenterol. 95:114–121. 2000.
|
|
31
|
Yamaguchi H, Goldenring JR, Kaminishi M
and Lee JR: Association of spasmolytic polypeptide-expressing
metaplasia with carcinogen administration and oxyntic atrophy in
rats. Lab Invest. 82:1045–1052. 2002.
|
|
32
|
Halldórsdóttir AM, Sigurdardóttrir M,
Jónasson JG, Oddsdóttir M, Magnússon J, Lee JR and Goldenring JR:
Spasmolytic polypeptide-expressing metaplasia (SPEM) associated
with gastric cancer in iceland. Dig Dis Sci. 48:431–441. 2003.
|
|
33
|
Yamaguchi H, Goldenring JR, Kaminishi M
and Lee JR: Identification of spasmolytic polypeptide expressing
metaplasia (SPEM) in remnant gastric cancer and surveillance
postgastrectomy biopsies. Dig Dis Sci. 47:573–578. 2002.
|
|
34
|
Goldenring JR and Nomura S:
Differentiation of the gastric mucosa III. Animal models of oxyntic
atrophy and metaplasia. Am J Physiol Gastrointest Liver Physiol.
291:G999–G1004. 2006.
|
|
35
|
Malfertheiner P, Megraud F, O'Morain CA,
Gisbert JP, Kuipers EJ, Axon AT, Bazzoli F, Gasbarrini A, Atherton
J, Graham DY, et al: Management of Helicobacter pylori
infection-the Maastricht V/Florence Consensus Report. Gut. 66:6–30.
2017.
|
|
36
|
Kuo HY, Chang WL, Yeh YC, Cheng HC, Tsai
YC, Wu CT, Lin SH, Yang HB, Lu CC and Sheu BS: Spasmolytic
polypeptide-expressing metaplasia associated with higher
expressions of miR-21, 155, and 223 can be regressed by
Helicobacter pylori eradication in the gastric cancer familial
relatives. Helicobacter. 24:e125782019.
|
|
37
|
Ogawa M, Nomura S, Car BD and Goldenring
JR: Omeprazole treatment ameliorates oxyntic atrophy induced by
DMP-777. Dig Dis Sci. 51:431–439. 2006.
|
|
38
|
Nam KT, Lee HJ, Sousa JF, Weis VG, O'Neal
RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM Jr, et al:
Mature chief cells are cryptic progenitors for metaplasia in the
stomach. Gastroenterology. 139:2028–2037.e9. 2010.
|
|
39
|
Weis VG, Sousa JF, LaFleur BJ, Nam KT,
Weis JA, Finke PE, Ameen NA, Fox JG and Goldenring JR:
Heterogeneity in mouse spasmolytic polypeptide-expressing
metaplasia lineages identifies markers of metaplastic progression.
Gut. 62:1270–1279. 2013.
|
|
40
|
Houghton J, Stoicov C, Nomura S, Rogers
AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR and Wang TC:
Gastric cancer originating from bone marrow-derived cells. Science.
306:1568–1571. 2004.
|
|
41
|
Goldenring JR, Ray GS, Coffey RJ, Meunier
PC, Haley PJ, Barnes TB and Car BD: Reversible drug-induced oxyntic
atrophy in rats. Gastroenterology. 118:1080–1093. 2000.
|
|
42
|
Nomura S, Yamaguchi H, Ogawa M, Wang TC,
Lee JR and Goldenring JR: Alterations in gastric mucosal lineages
induced by acute oxyntic atrophy in wild-type and gastrin-deficient
mice. Am J Physiol Gastrointest Liver Physiol. 288:G362–G375.
2005.
|
|
43
|
Nozaki K, Ogawa M, Williams JA, Lafleur
BJ, Ng V, Drapkin RI, Mills JC, Konieczny SF, Nomura S and
Goldenring JR: A molecular signature of gastric metaplasia arising
in response to acute parietal cell loss. Gastroenterology.
134:511–522. 2008.
|
|
44
|
Nam KT, Varro A, Coffey RJ and Goldenring
JR: Potentiation of oxyntic atrophy-induced gastric metaplasia in
amphiregulin-deficient mice. Gastroenterology. 132:1804–1819.
2007.
|
|
45
|
Muthupalani S, Ge Z, Joy J, Feng Y, Dobey
C, Cho HY, Langenbach R, Wang TC, Hagen SJ and Fox JG: Muc5ac null
mice are predisposed to spontaneous gastric antro-pyloric
hyperplasia and adenomas coupled with attenuated H. pylori-induced
corpus mucous metaplasia. Lab Invest. 99:1887–1905. 2019.
|
|
46
|
Busada JT, Ramamoorthy S, Cain DW, Xu X,
Cook DN and Cidlowski JA: Endogenous glucocorticoids prevent
gastric metaplasia by suppressing spontaneous inflammation. J Clin
Invest. 129:1345–1358. 2019.
|
|
47
|
Biswas SK and Mantovani A: Macrophage
plasticity and interaction with lymphocyte subsets: Cancer as a
paradigm. Nat Immunol. 11:889–896. 2010.
|
|
48
|
Hanna RN, Cekic C, Sag D, Tacke R, Thomas
GD, Nowyhed H, Herrley E, Rasquinha N, McArdle S, Wu R, et al:
Patrolling monocytes control tumor metastasis to the lung. Science.
350:985–990. 2015.
|
|
49
|
Papierska L and Rabijewski M: Delay in
diagnosis of adrenal insufficiency is a frequent cause of adrenal
crisis. Int J Endocrinol. 2013:4823702013.
|
|
50
|
Puar TH, Stikkelbroeck NM, Smans LC,
Zelissen PM and Hermus AR: Adrenal crisis: still a deadly event in
the 21st century. Am J Med. 129:339 e1–9. 2016.
|
|
51
|
Meyer AR, Engevik AC, Madorsky T, Belmont
E, Stier MT, Norlander AE, Pilkinton MA, McDonnell WJ, Weis JA,
Jang B, et al: Group 2 innate lymphoid cells coordinate damage
response in the stomach. Gastroenterology. 159:2077–2091.e8.
2020.
|
|
52
|
Meyer AR and Goldenring GR: Injury,
repair, inflammation and metaplasia in the stomach. J Physiol.
596:3861–3867. 2018.
|
|
53
|
Goldenring JR, Nam KT, Wang TC, Mills JC
and Wright NA: Spasmolytic polypeptide-expressing metaplasia and
intestinal metaplasia: Time for reevaluation of metaplasias and the
origins of gastric cancer. Gastroenterology. 138:2207–2210.e1.
2010.
|
|
54
|
Hayakawa Y and Wang TC: Isthmus
Progenitors, not chief cells, are the likely origin of metaplasia
in eR1-CreERT; LSL-KrasG12D Mice. Gastroenterology.
152:2078–2079. 2017.
|
|
55
|
Hayakawa Y, Fox JG and Wang TC: The
origins of gastric cancer from gastric stem cells: Lessons from
mouse models. Cell Mol Gastroenterol Hepatol. 3:331–338. 2017.
|
|
56
|
Nomura S, Baxter T, Yamaguchi H, Leys C,
Vartapetian AB, Fox JG, Lee JR, Wang TC and Goldenring JR:
Spasmolytic polypeptide expressing metaplasia to preneoplasia in H.
felis-infected mice. Gastroenterology. 127:582–594. 2004.
|
|
57
|
Kinoshita H, Hayakawa Y, Niu Z, Konishi M,
Hata M, Tsuboi M, Hayata Y, Hikiba Y, Ihara S, Nakagawa H, et al:
Mature gastric chief cells are not required for the development of
metaplasia. Am J Physiol Gastrointest Liver Physiol. 314:G583–G596.
2018.
|
|
58
|
Hata M, Kinoshita H, Hayakawa Y, Konishi
M, Tsuboi M, Oya Y, Kurokawa K, Hayata Y, Nakagawa H, Tateishi K,
et al: GPR30-Expressing gastric chief cells do not dedifferentiate
but are eliminated via PDK-Dependent cell competition during
development of metaplasia. Gastroenterology. 158:1650–1666 e15.
2020.
|
|
59
|
Nam KT, O'Neal RL, Coffey RJ, Finke PE,
Barker N and Goldenring JR: Spasmolytic polypeptide-expressing
metaplasia (SPEM) in the gastric oxyntic mucosa does not arise from
Lgr5-expressing cells. Gut. 61:1678–1685. 2012.
|
|
60
|
Meyer AR, Engevik AC, Willet SG, Williams
JA, Zou Y, Massion PP, Mills JC, Choi E and Goldenring JR:
Cystine/Glutamate Antiporter (xCT) is required for chief cell
plasticity after gastric injury. Cell Mol Gastroenterol Hepatol.
8:379–405. 2019.
|
|
61
|
Lee A, O'Rourke J, De Ungria MC, Robertson
B, Daskalopoulos G and Dixon MF: A standardized mouse model of
Helicobacter pylori infection: Introducing the Sydney strain.
Gastroenterology. 112:1386–1397. 1997.
|
|
62
|
Watanabe T, Tada M, Nagai H, Sasaki S and
Nakao M: Helicobacter pylori infection induces gastric cancer in
Mongolian gerbils. Gastroenterology. 115:642–648. 1998.
|
|
63
|
Lee JR, Baxter TM, Yamaguchi H, Wang TC,
Goldenring JR and Anderson MG: Differential protein analysis of
spasomolytic polypeptide expressing metaplasia using laser capture
microdissection and two-dimensional difference gel electrophoresis.
Appl Immunohistochem Mol Morphol. 11:188–193. 2003.
|
|
64
|
Yoshizawa N, Takenaka Y, Yamaguchi H,
Tetsuya T, Tanaka H, Tatematsu M, Nomura S, Goldenring JR and
Kaminishi M: Emergence of spasmolytic polypeptide-expressing
metaplasia in Mongolian gerbils infected with Helicobacter pylori.
Lab Invest. 87:1265–1276. 2007.
|
|
65
|
El-Zimaity HM, Ramchatesingh J, Saeed MA
and Graham DY: Gastric intestinal metaplasia: Subtypes and natural
history. J Clin Pathol. 54:679–683. 2001.
|
|
66
|
Matsukura N, Kinebuchi M, Kawachi T, Sato
S and Sugimura T: Quantitative measurement of intestinal marker
enzymes in intestinal metaplasia from human stomach with cancer.
Gan. 70:509–513. 1979.
|
|
67
|
Ricardo SD, van Goor H and Eddy AA:
Macrophage diversity in renal injury and repair. J Clin Invest.
118:3522–3530. 2008.
|
|
68
|
Mills CD, Kincaid K, Alt JM, Heilman MJ
and Hill AM: M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J
Immunol. 164:6166–6173. 2000.
|
|
69
|
Teal E, Dua-Awereh M, Hirshorn ST and
Zavros Y: Role of metaplasia during gastric regeneration. Am J
Physiol Cell Physiol. 319:C947–C954. 2020.
|
|
70
|
Petersen CP, Meyer AR, De Salvo C, Choi E,
Schlegel C, Petersen A, Engevik AC, Prasad N, Levy SE, Peebles RS,
et al: A signalling cascade of IL-33 to IL-13 regulates metaplasia
in the mouse stomach. Gut. 67:805–817. 2018.
|
|
71
|
De Salvo C, Pastorelli L, Petersen CP,
Buttò LF, Buela KA, Omenetti S, Locovei SA, Ray S, Friedman HR,
Duijser J, et al: Interleukin 33 triggers early
eosinophil-dependent events leading to metaplasia in a chronic
model of gastritis-prone mice. Gastroenterology. 160:302–316 e7.
2021.
|
|
72
|
Xu X, Cheng J, Luo S, Gong X, Huang D, Xu
J, Qian Y, Wan X and Zhou H: Deoxycholic acid-stimulated
macrophage-derived exosomes promote spasmolytic
polypeptide-expressing metaplasia in the stomach. Biochem Biophys
Res Commun. 524:649–655. 2020.
|
|
73
|
Park H, Li Z, Yang XO, Chang SH, Nurieva
R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q and Dong C: A distinct
lineage of CD4 T cells regulates tissue inflammation by producing
interleukin 17. Nat Immunol. 6:1133–1141. 2005.
|
|
74
|
Harrington LE, Hatton RD, Mangan PR,
Turner H, Murphy TL, Murphy KM and Weaver CT: Interleukin
17-producing CD4+ effector T cells develop via a lineage distinct
from the T helper type 1 and 2 lineages. Nat Immunol. 6:1123–1132.
2005.
|
|
75
|
Onishi RM and Gaffen SL: Interleukin-17
and its target genes: Mechanisms of interleukin-17 function in
disease. Immunology. 129:311–321. 2010.
|
|
76
|
Bockerstett KA, Osaki LH, Petersen CP, Cai
CW, Wong CF, Nguyen TM, Ford EL, Hoft DF, Mills JC, Goldenring JR
and DiPaolo RJ: Interleukin-17A promotes parietal cell atrophy by
inducing apoptosis. Cell Mol Gastroenterol Hepatol. 5:678–690 e1.
2018.
|
|
77
|
Togawa S, Joh T, Itoh M, Katsuda N, Ito H,
Matsuo K, Tajima K and Hamajima N: Interleukin-2 gene polymorphisms
associated with increased risk of gastric atrophy from helicobacter
pylori infection. Helicobacter. 10:172–178. 2005.
|
|
78
|
Sugimoto M, Yamaoka Y and Furuta T:
Influence of interleukin polymorphisms on development of gastric
cancer and peptic ulcer. World J Gastroenterol. 16:1188–1200.
2010.
|
|
79
|
Yamaoka Y, Kodama T, Kita M, Imanishi J,
Kashima K and Graham DY: Relation between clinical presentation,
Helicobacter pylori density, interleukin 1beta and 8 production,
and cagA status. Gut. 45:804–811. 1999.
|
|
80
|
Lee C, Lee H, Hwang SY, Moon CM and Hong
SN: IL-10 Plays a pivotal role in tamoxifen-induced spasmolytic
polypeptide-expressing metaplasia in gastric mucosa. Gut Liver.
11:789–797. 2017.
|
|
81
|
Maloum F, Allaire JM, Gagné-Sansfaçon J,
Roy E, Belleville K, Sarret P, Morisset J, Carrier JC, Mishina Y,
Kaestner KH and Perreault N: Epithelial BMP signaling is required
for proper specification of epithelial cell lineages and gastric
endocrine cells. Am J Physiol Gastrointest Liver Physiol.
300:G1065–G1079. 2011.
|
|
82
|
Bleuming SA, He XC, Kodach LL, Hardwick
JC, Koopman FA, Ten Kate FJ, van Deventer SJ, Hommes DW,
Peppelenbosch MP, Offerhaus GJ, et al: Bone morphogenetic protein
signaling suppresses tumorigenesis at gastric epithelial transition
zones in mice. Cancer Res. 67:8149–8155. 2007.
|
|
83
|
Oshima H, Itadani H, Kotani H, Taketo MM
and Oshima M: Induction of prostaglandin E2 pathway promotes
gastric hamartoma development with suppression of bone
morphogenetic protein signaling. Cancer Res. 69:2729–2733.
2009.
|
|
84
|
Auclair BA, Benoit YD, Rivard N, Mishina Y
and Perreault N: Bone morphogenetic protein signaling is essential
for terminal differentiation of the intestinal secretory cell
lineage. Gastroenterology. 133:887–896. 2007.
|
|
85
|
Oshima H and Oshima M: Mouse models of
gastric tumors: Wnt activation and PGE2 induction. Pathol Int.
60:599–607. 2010.
|
|
86
|
Liu Z, Demitrack ES, Keeley TM, Eaton KA,
El-Zaatari M, Merchant JL and Samuelson LC: IFNү contributes to the
development of gastric epithelial cell metaplasia in Huntingtin
interacting protein 1 related (Hip1r)-deficient mice. Lab Invest.
92:1045–1057. 2012.
|
|
87
|
Wang TC, Dangler CA, Chen D, Goldenring
JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ
and Fox JG: Synergistic interaction between hypergastrinemia and
Helicobacter infection in a mouse model of gastric cancer.
Gastroenterology. 118:36–47. 2000.
|
|
88
|
Mohammadi M, Czinn S, Redline R and Nedrud
J: Helicobacter-specific cell-mediated immune responses display a
predominant Th1 phenotype and promote a delayed-type
hypersensitivity response in the stomachs of mice. J Immunol.
156:47291996.
|
|
89
|
Roth KA, Kapadia SB, Martin SM and Lorenz
RG: Cellular immune responses are essential for the development of
helicobacter felis-Associated gastric pathology. J Immunol.
163:14901999.
|
|
90
|
Srivastava S, Huang KK, Rebbani K, Das K,
Fazreen Z, Yeoh KG, Tan P and The M: An LCM-based genomic analysis
of SPEM, gastric cancer and pyloric gland adenoma in an Asian
cohort. Mod Pathol. 33:2075–2086. 2020.
|
|
91
|
Hernandez C, Huebener P and Schwabe RF:
Damage-associated molecular patterns in cancer: A double-edged
sword. Oncogene. 35:5931–5941. 2016.
|
|
92
|
Otani K, Tanigawa T, Watanabe T, Nadatani
Y, Sogawa M, Yamagami H, Shiba M, Watanabe K, Tominaga K, Fujiwara
Y and Arakawa T: Toll-like receptor 9 signaling has
anti-inflammatory effects on the early phase of Helicobacter
pylori-induced gastritis. Biochem Biophys Res Commun. 426:342–349.
2012.
|
|
93
|
Varga MG, Piazuelo MB, Romero-Gallo J,
Delgado AG, Suarez G, Whitaker ME, Krishna US, Patel RV, Skaar EP,
Wilson KT, et al: TLR9 activation suppresses inflammation in
response to Helicobacter pylori infection. Am J Physiol
Gastrointest Liver Physiol. 311:G852–G858. 2016.
|
|
94
|
Varga MG, Shaffer CL, Sierra JC, Suarez G,
Piazuelo MB, Whitaker ME, Romero-Gallo J, Krishna US, Delgado A,
Gomez MA, et al: Pathogenic Helicobacter pylori strains translocate
DNA and activate TLR9 via the cancer-associated cag type IV
secretion system. Oncogene. 35:6262–6269. 2016.
|
|
95
|
Wang X, Xue L, Yang Y, Xu L and Zhang G:
TLR9 promoter polymorphism is associated with both an increased
susceptibility to gastric carcinoma and poor prognosis. PLoS One.
8:e657312013.
|
|
96
|
Min J, Han TS, Sohn Y, Shimizu T, Choi B,
Bae SW, Hur K, Kong SH, Suh YS, Lee HJ, et al: microRNA-30a
arbitrates intestinal-type early gastric carcinogenesis by directly
targeting ITGA2. Gastric Cancer. 23:600–613. 2020.
|
|
97
|
Riera KM, Jang B, Min J, Roland JT, Yang
Q, Fesmire WT, Camilleri-Broet S, Ferri L, Kim WH, Choi E and
Goldenring JR: Trop2 is upregulated in the transition to dysplasia
in the metaplastic gastric mucosa. J Pathol. 251:336–347. 2020.
|
|
98
|
Fox JG and Wang TC: Inflammation, atrophy,
and gastric cancer. J Clin Invest. 117:60–69. 2007.
|
|
99
|
Correa P: Human gastric carcinogenesis: A
multistep and multifactorial process-First American Cancer Society
Award Lecture on Cancer Epidemiology and Prevention. Cancer Res.
52:6735–6740. 1992.
|
|
100
|
Thompson MP and Kurzrock R: Epstein-Barr
virus and cancer. Clin Cancer Res. 10:803–821. 2004.
|
|
101
|
Murphy G, Pfeiffer R, Camargo MC and
Rabkin CS: Meta-analysis shows that prevalence of Epstein-Barr
virus-positive gastric cancer differs based on sex and anatomic
location. Gastroenterology. 137:824–833. 2009.
|
|
102
|
Cancer Genome Atlas Research Network:
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature. 513:202–209. 2014.
|
|
103
|
Lee JH, Kim SH, Han SH, An JS, Lee ES and
Kim YS: Clinicopathological and molecular characteristics of
Epstein-Barr virus-associated gastric carcinoma: A meta-analysis. J
Gastroenterol Hepatol. 24:354–365. 2009.
|
|
104
|
Zhang Y, Chen JN, Dong M, Zhang ZG, Zhang
YW, Wu JY, Du H, Li HG, Huang Y and Shao CK: Clinical significance
of spasmolytic polypeptide-expressing metaplasia and intestinal
metaplasia in Epstein-Barr virus-associated and Epstein-Barr
virus-negative gastric cancer. Hum Pathol. 63:128–138. 2017.
|
|
105
|
Gessner R and Tauber R: Intestinal cell
adhesion molecules: liver-intestine cadherin. Ann NY Acad Sci.
915:136–143. 2000.
|
|
106
|
Lee HJ, Nam KT, Park HS, Kim MA, Lafleur
BJ, Aburatani H, Yang HK, Kim WH and Goldenring JR: Gene expression
profiling of metaplastic lineages identifies CDH17 as a prognostic
marker in early stage gastric cancer. Gastroenterology.
139:213–725.e3. 2010.
|
|
107
|
O'Neal RL, Nam KT, LaFleur BJ, Barlow B,
Nozaki K, Lee HJ, Kim WH, Yang HK, Shi C, Maitra A, et al: Human
epididymis protein 4 is up-regulated in gastric and pancreatic
adenocarcinomas. Hum Pathol. 44:734–742. 2013.
|
|
108
|
Sousa JF, Ham AJ, Whitwell C, Nam KT, Lee
HJ, Yang HK, Kim WH, Zhang B, Li M, LaFleur B, et al: Proteomic
profiling of paraffin-embedded samples identifies
metaplasia-specific and early-stage gastric cancer biomarkers. Am J
Pathol. 181:1560–1572. 2012.
|
|
109
|
Merchant JL and Ding L: Hedgehog signaling
links chronic inflammation to gastric cancer precursor lesions.
Cell Mol Gastroenterol Hepatol. 3:201–210. 2017.
|
|
110
|
Bockerstett KA, Lewis SA, Noto CN, Ford
EL, Saenz JB, Jackson NM, Ahn TH, Mills JC and DiPaolo RJ:
Single-Cell transcriptional analyses identify lineage-specific
epithelial responses to inflammation and metaplastic development in
the gastric corpus. Gastroenterology. 159:2116–2129 e4. 2020.
|
|
111
|
Lee SH, Jang B, Min J, Contreras-Panta EW,
Presentation KS, Delgado AG, Piazuelo MB, Choi E and Goldenring JR:
Up-regulation of aquaporin 5 defines spasmolytic
polypeptide-expressing metaplasia and progression to incomplete
intestinal metaplasia. Cell Mol Gastroenterol Hepatol. 13:199–217.
2022.
|