Introduction
Renal cancer accounts for 5% of all cancer and ranks
as the sixth most prevalent type of cancer in male patients. In
female patients, it contributes to 3% of malignancies and stands as
the tenth most popular form of tumor (1,2).
Renal cell carcinoma (RCC) accounts for >90% of kidney tumors
(2,3). Depending on population surveys, a
5-year survival rate was observed in 70% of patients diagnosed with
regional cancer. However, for those with metastatic RCC, this rate
drops down to 13% (4). Clear cell
RCC (ccRCC) is the most common histological subtype of RCC,
accounting for 75% of cases, which develops in the epithelium of
kidney tubules and can metastasize to different organs (5).
The inactivation of the tumor suppressor gene Von
Hippel Lindau (VHL) has a key role in the pathogenesis of clear
cell carcinoma (6). It was
demonstrated that >90% of patients with ccRCC experience a loss
of VHL heterozygosity and inactivating mutations of VHL are
detected in 50-65% of instances (7,8).
Reducing the level of VHL proteins directly promotes an increase in
the expression of both hypoxia-inducible factors (HIFs) including
HIF-1A and HIF-2A (9). These
factors activate various target genes in the tumor microenvironment
which are stimulating tumor cell survival, cell proliferation,
angiogenesis, metabolism of sugar and tumor metastasis (10-12).
Targeting HIFs or downstream effector molecules in the VHL/HIF
pathway (such as VEGFs) has been used for three decades in the
treatment of disease outcomes in advanced patients with ccRCC
(13).
Sunitinib is one of the earliest approved
multi-targeted tyrosine kinase inhibitors (TKIs) that have been
extensively used as first-line treatment for metastatic ccRCC
(14). TKIs act by inhibiting the
kinase activity of various receptors and are still used in therapy
(15). Notably, despite its
demonstrated efficacy in treating advanced RCC, a substantial
proportion of patients develop primary resistance or acquired
resistance to sunitinib within 6-15 months of treatment (16). BAY-876 was previously identified as
a new-generation inhibitor of glucose transporter 1 (GLUT1) in
ovarian cancer (17); however,
since its discovery, there have been no studies investigating the
effect of BAY-876 as GLUTI in ccRCC.
Hydrogen sulfide (H2S) is a gaseous
transmitter found in mammalian tissues; along with nitric oxide
(NO) and carbon monoxide (CO), it has a variety of biological
functions (18,19). Other studies demonstrated that
H2S gas has a dual effect on cancerous diseases, acting
as both a pro- and antitumor agent (20,21).
Sodium hydrosulfide (NaSH) salt is a solid analog of H2S
gas that provides immediate and direct access to biologically
significant forms of sulfide. NaSH has been used extensively as an
exogenous delivery method for H2S to evaluate its
therapeutic potential (22). To
the best of the authors' knowledge, the effects of NaSH and BAY-876
on RCC remain unclear and a significant number of patients with
ccRCC have demonstrated a lack of response, recurrence, or
resistance to sunitinib. For these reasons, the present study aimed
to investigate the effects of NaSH and BAY-876 alone and in
combination with sunitinib on ccRCC cells. The evaluation of
genotypic variations and polymorphisms in the VHL and kinase insert
domain receptor (KDR) genes in patients with ccRCC was another aim
of the present study.
Materials and methods
Cell line
The human metastatic ccRCC cell line (786-O) was
obtained from the American Type Culture Collection (cat. no.
CRL-1932). The cells were cultured in RPMI-1640 medium (Thermo
Fisher Scientific, Inc.; cat. no. 31800089) which was supplemented
with 10% fetal bovine serum (FBS) and 1% of antibiotic (consisting
of 10,000-unit penicillin, 10 mg streptomycin and 25 µg
amphotericin; Sigma Aldrich; Merck KGaA; cat. no. A5955). Cells
were split every 2-3 days to maintain their density and maintained
at 37˚C in a humidified incubator with 5% CO2 and 95% air
(RS Biotech Galaxy Model R+). Following an adaptation step of three
passages, the experiments were conducted within passages 11 to 16
of 786-O cells (23).
Determining IC50
To determine the IC50 of sunitinib
(MedChemExpress; cat. no. HY-10255A), BAY-876 (MedChemExpress; cat.
no. HY-100017) and NaSH on 786-O viability, cells were seeded in a
96-well culture plate (4x103 cell/well) and incubated at
37˚C in a humidified incubator with 5% CO2. After 24 h,
the cells were treated with various concentrations of sunitinib
(range, 1-100 µM), BAY-876 (range, 10-1,000 µM) and NaSH (range,
10-103 µM) in quadruplicate for each concentration. The
plates were incubated for 24, 48 and 72 h at 37˚C with 5%
CO2. For BAY-876 another plate was incubated for 96 h.
After each incubation interval, the culture media of the plate
wells were changed with new media and then the cell cytotoxicity
was measured by using Cell Counting Kit-8 (CCK-8; MedChemExpress;
cat. no. HY-K0301) (24) according
to the manufacturer's instruction. Briefly, 10 µM of the CCK-8
reagent was added to each well, incubated for 2-4 h at 37˚C and
then the absorbance was measured at 540 nm by using an ELIZA reader
(BioTek Instruments, Inc.). Each experiment was carried out three
times.
Drug combinations
The IC50 of sunitinib after 72 h
(5.26x10-6 M), BAY-876 after 96 h (53.56x10-6
M) and NaSH after 72 h (692x10-6 M) were used to
determine the cytotoxicity of the combination of drugs against
786-O cells. Briefly, 4x103 cells/well were seeded in 96
well plates and incubated at 37˚C with 5% CO2. After 24
h incubation, the cells were exposed to the IC50 of each
drug alone and a combination of sunitinib with BAY-876, sunitinib
with NaSH and BAY-876 with NaSH. Following incubation for 24 and 48
h at 37˚C and 5% CO2, cell cytotoxicity was measured by
using the CCK-8 kit.
Statistical analysis
Data analysis was conducted via GraphPad Prism 9
Software (Dotmatics). Non-linear regression was employed to
establish the IC50 of medications, while ordinary
one-way ANOVA with Tukey's multiple comparisons test was performed
to assess the statistically significant differences in the
anticancer activity between various agents and untreated cells. The
data were reported as the mean value ± standard error (SE) of the
difference. P<0.05 was considered to indicate a statistically
significant difference.
Formalin-fixed paraffin-embedded
tissues of patients with ccRCC
A total of 30 formalin-fixed paraffin-embedded
tissues (FFPE) of patients with ccRCC were collected form PAR and
Rzgary teaching hospitals (Erbil, Iraq) between July 2022 and May
2023. After histological confirmation of ccRCC through hematoxylin
and eosin (H&E) staining, the blocks were collected for
extraction of DNA. The approval (approval no. 45/90) of the present
study was granted by the Human Ethics Committee of the College of
Education of Salahaddin University-Erbil (Erbil, Iraq). The study
was conducted according to the criteria set by The Declaration of
Helsinki (October 2013) and signed informed consent forms were
obtained from the participants approving the use of their tissues
for investigation. DNA samples were extracted from the FFPE
tissues. Clinicopathological characteristics of patients (including
sex and age distribution) are presented in Table I.
 | Table IClinicopathological characteristics
of patients with clear cell renal cell carcinoma. |
Table I
Clinicopathological characteristics
of patients with clear cell renal cell carcinoma.
| Clinicopathological
characteristics | Percentage (%) |
|---|
| Sex | |
|
Male | 44.40 |
|
Female | 56.60 |
| Age, years | |
|
>50 | 50 |
|
51-60 | 20 |
|
61-70 | 20 |
|
<71 | 10 |
| Degree of
differentiation | |
|
Well-differentiated
tumor | 43.33 |
|
Moderately
differentiated tumor | 50 |
|
Poorly
differentiated | 6.66 |
| TNM staging | |
|
Stage I | |
|
pT1a
N0 Mx | 50 |
|
pT1b
N0 Mx | 26.60 |
|
Stage
II | |
|
pT2
N0 Mx | 3.33 |
|
Stage
III | |
|
pT1b
N1 Mx | 6.33 |
|
pT3a
Nx Mx | 3.66 |
|
pT3a
N0 Mx | 6.33 |
|
pT3
N1 Mx | 3.33 |
Stage assessment of ccRCC
The TNM cancer staging system provides significant
predictors of prognosis in RCC. The present study used the eighth
edition (TNM8) of the American Joint Committee on Cancer (AJCC) and
the Union for International Cancer Control (UICC) for ccRCC tumor
staging (25,26). In TNM, the T refers to the size and
regional extension of the primary tumor, the N refers to the tumor
involvement in the regional lymph nodes and the M refers to
metastasis or spread of the tumor to other parts of the body away
from the original location (27).
DNA extraction from FFPE tissues
Genomic DNA extraction from FFPE was performed using
the QIAamp DNA FFPE Tissue kit (Qiagen GmbH; cat. no. 56404)
according to the manufacturer's protocol. Briefly, 5-µm thick
ribbon sections were created from the paraffin blocks, followed by
deparaffination by xylene. The cells were lysed with lysis buffer
and proteinase k. The DNA bound to the membrane and the
contaminants were flowing through the membrane. Finally, pure DNA
was eluted from the membrane with an elution buffer. The eluted DNA
was quantified based on the absorbance (A) measured at 260 nm
(A260) and purity was estimated based on the A260/A280 ratio using
a Nanodrop (Thermo Fisher Scientific, Inc.). An elution buffer was
used as a blank for the Nanodrop instrument. The assessment of DNA
purity relied on an ideal A260/A280 ratio of 1.8(28).
Genotype determination
The current study examined variants of each VHL and
KDR gene. Based on the results of a previous study, exon 1 of VHL
(29) and exon 13 of KDR genes
were chosen for the current study. Initially, the purified DNA
underwent an independent process of amplification. For this
purpose, a pre-prepared master mix (including dNTP,
MgCl2, Taq DNA polymerase and PCR reaction buffer;
Promega Corporation) and the following primer pairs were used: VHL
forward, 5'-GTCTGGATCGCGGAGGGAAT-3' and reverse,
5'-GGACTGCGATTGCAGAAGAT-3'; KDR forward, 5'-CGTGTCTTTGTGGTGCACTG-3'
and reverse, 5'-CTAGGACTGTGAGCTGCCTG-3'. The thermocycling
conditions used for PCR were: Initial denaturation at 95˚C for 5
min; 35 cycles of denaturation at 95˚C for 30 sec, annealing at
61˚C for 30 sec and extension at 72˚C for 1 min; final elongation
at 72˚C for 6 min and the samples were maintained at 4˚C.
The PCR products were separated by 1.5% agarose gel
electrophoresis. DNA ladder of 100 base pairs (GeneDireX, Inc.;
cat. no. DM003-R500) was used for band composition as shown in
Fig. S1. Before pouring it into
the tray, the gel was mixed with Safe gel stain Dye (ADD BIO) and
then it was visualized using the UV Transilluminator UST-20M-8K
(Biostep GmbH). Following the PCR protocol, the products were
delivered to the cytogenetic laboratory-Zheen International
Hospital-Erbil-Iraq for sequencing performed on the Automatic 3130
Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.) using the same forward primers for each exon in the genes.
The sequencing data were deposited into the GenBank database
(https://www.ncbi.nlm.nih.gov/genbank/; accession no.
OR294044 and OR339875). Analysis and interpretation of the Sanger
sequencing data were performed using the Mutation Surveyor software
package 5.1 (SoftGenetics, LLC) by comparing the sequencing results
with reference genes in public databases, including ClinVar, dbSNP,
gnomAD and COSMIC (30).
Database's gene mutation retrieval and
gene interactions
Genome aggregation database (gnomAD) version 3.1.2
(https://gnomad.broadinstitute.org)
was used for mutation retrieval of selected genes. This population
referencing database currently is one of the most powerful tools
that provides information about genetic variation and gene
interpretation found in the human population. This resource is
developed by worldwide investigators who collect and integrate both
genome and exome data from massive sequencing programs. The
database nowadays is widely used in clinical genetics and genomic
research (31).
All recorded gene mutations of chosen genes (VHL and
KDR) in RCC were retrieved from both the Mutagene database
(https://www.ncbi.nlm.nih.gov/research/mutagene) and
Catalog of Somatic Mutation of Cancer (COSMIC v98, released
23-MAY-23; https://cancer.sanger.ac.uk/cosmic). The Mutagene
database contains 441 RRC samples with 33,063 gene mutations. Based
on Bscore thresholds, Mutagene is used to determine the
cancer driver, potential cancer driver and passenger mutations of
selected genes. COSMIC, which is readily available to researchers
globally, is a thorough database encompassing somatic mutations in
cancer. It provides extensive information about mutation location,
frequency and impact, along with clinical and pathological data. By
utilizing this database, researchers can identify prevalent
mutations and other genetic changes that potentially play a role in
the development or advancement of cancer. Moreover, these findings
may offer potential targets for therapeutic interventions (32).
In addition, the interaction of VHL and KDR genes in
RCC is predicted through the GeneMANIA anticipation tool
(https://genemania.org/). Based on genetic
connection, physical protein-protein interaction and co-expression
pattern, this public online database can provide gene interaction.
By examining the anticipated associations among these two genes,
valuable insights into their participation in cellular processes
and disease pathways can be gained. This, in turn, could pave the
way for the identification of fresh therapeutic targets or
diagnostic biomarkers. GeneMANIA through anticipation of gene
interaction will lead to determining the function of genes,
uncovering gene interaction in specific cellular assays (33) and identifying potential gene
candidates for further study.
Results
Anticancer activity of sunitinib,
BAY-876 and NaSH
To determine the in vitro cytotoxic effect of
sunitinib, BAY-876 and NaSH, dose-dependent viability studies were
performed to find the appropriate concentration of each drug that
can eliminate 50% (IC50) of 786-O cells by using the
CCK-8 kit. The IC50s for sunitinib were found to be
9.85x106, 9.1x10-6 and 5.26x10-6 M
at 24, 48 and 72 h, respectively (Fig.
1A), with no significant difference among the measured
timepoints. The IC50 for BAY-876 was reached only after
4 days of treatment and it was 53.56x10-6 M (Fig. 1B). Furthermore, the present study
also revealed that the IC50 for NaSH as an exogenous
donor of H2S after 24 and 48 h of treatment were equal
to 726x10-6 and 692x10-6 M, respectively
(Fig. 1C), with no significant
difference between the two timepoints.
 | Figure 1Time- and dose-dependent effect of
(A) sunitinib, (B) BAY-876 and (C) NaSH on viability of 786-O
cells. Cell viability (%) was expressed as the percentage of
treated cells versus log of concentration. Data are expressed as
the mean values ± standard error. The *, ■, ʈ, ⊗, Ψ and ᵶ represent
significant difference in the cytotoxicity effect of specific
concentration between 24 and 48, 48 and 72, 48 and 96, 24 and 96,
72 and 96 and 24-72 h of incubation respectively. ■,
ᵶP<0.05, ʈʈ, Ψ Ψ,
ᵶ ᵶP<0.01 and ***, ■ ■
■, ʈʈʈ, ⊗⊗⊗, ΨΨΨ,
ᵶᵶᵶP<0.001. |
In addition, the cytotoxic effect against 786-O
cells of several drug combinations was assessed. Sunitinib
IC50 after 3 days, BAY-IC50 after 4 days and
NaSH after 2 days were used alone and in combination for 24
(Fig. 2) and 48 h (Fig. 3). The results after 24 h
demonstrated that there were no significant differences in
cytotoxicity between sunitinib and BAY-876 treatments alone
compared with the control group; however, their combination
demonstrated statistically different cytotoxicity (P<0.001)
compared with the control group and each drug used
individually.
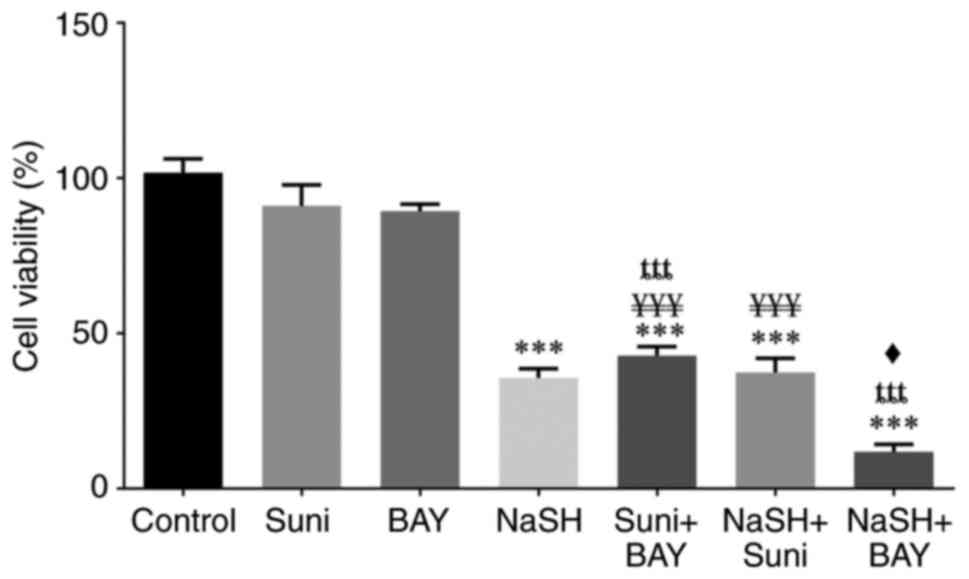
 | Figure 3Cytotoxicity effect of sunitinib,
BAY-876 and NaSH alone and in combination against 786-O cells after
48 h of treatment. The *, ¥, ȶ and ♦ represent statistical
differences from control, sunitinib, BAY-876, and NaSH
respectively. Each column represents the mean of four data; three
independent experiments were performed. ♦P<0.05,
**, ♦♦P<0.01 and ***, ¥¥¥,
ȶȶȶP<0.001. |
Regarding the cytotoxic effect of NaSH, NaSH alone
and in combination with either sunitinib or BAY-876 revealed a
statistically significant increase (P<0.001) in cytotoxicity
compared with the control group. The cytotoxicity of NaSH in
combination with BAY-876 was significantly higher (P<0.05)
compared with NaSH alone. However, there was no significant
difference between NaSH and sunitinib co-treatment and NaSH.
The outcomes of the drug combinations at 24 and 48 h
were comparable and the only exception was that there was a
statistically significant difference (P<0.05) between the NaSH
and sunitinib co-treatment and NaSH alone in reducing cell
viability.
Patient characteristics
In the current study, out of 30 patients with ccRCC,
17 were male (56.6%) and 13 were female (44.40%). Regarding the
patient age, 50 (50%) were under 50 years, 6 (20%) were between 51
and 60 years, 6 (20%) were between 61 and 70 years, and 3 (10%)
were >71 years old. The degree of cell differentiation varied
among patients with ccRCC. A large number of patients were grouped
as medium-differentiated (50%) and well-differentiated carcinomas
(43.33%), while poorly-differentiated carcinomas accounted only for
6.66% (Table I).
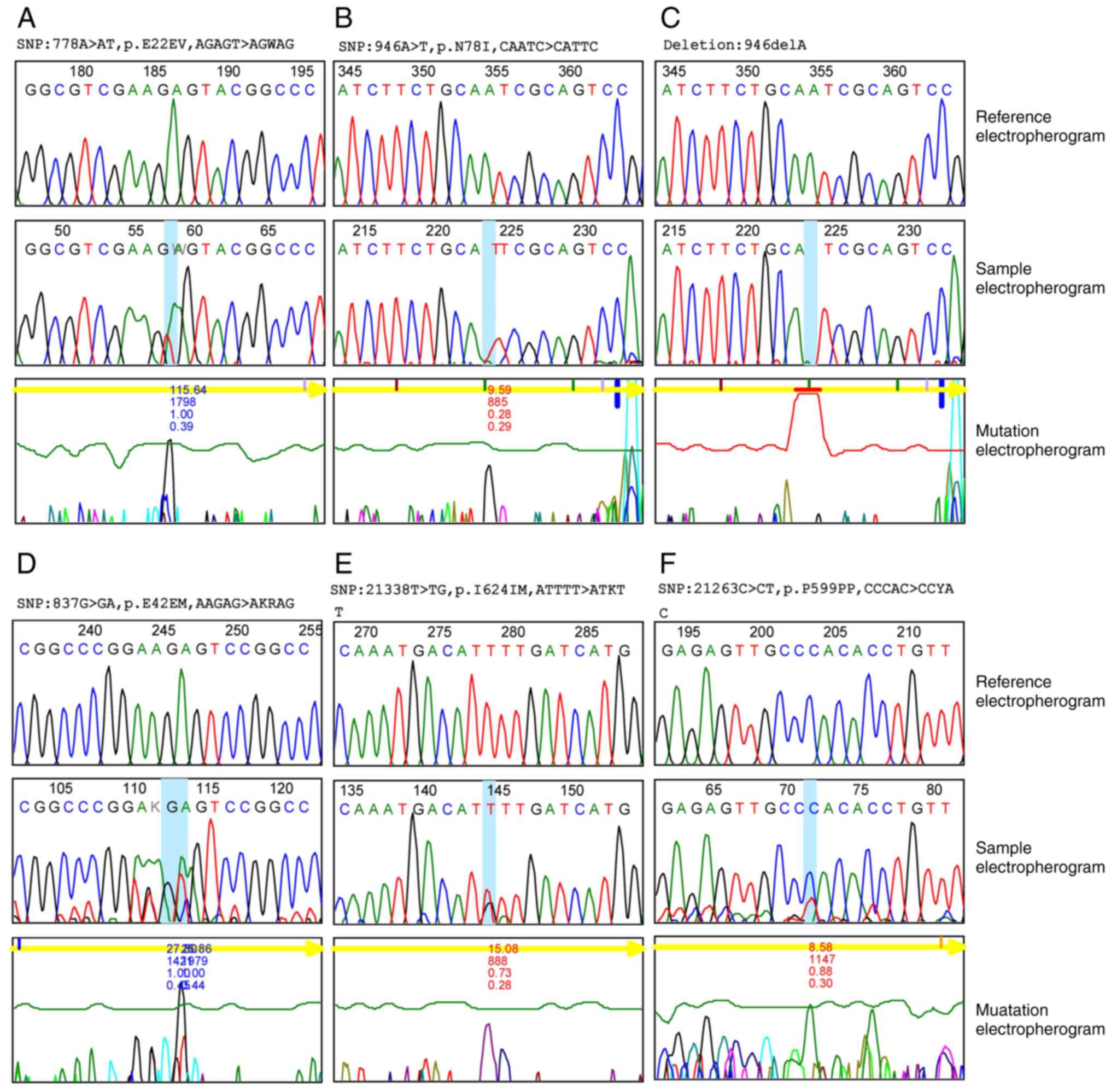
Mutation analysis
Single nucleotide polymorphism (SNP) was recorded in
47.7% of the VHL gene and 31.5% of the KDR gene (data not shown).
In exon 1 of the VHL gene of ccRCC, 36 SNP mutations were
identified, including 35 substitution mutations (A>AT, C>CT,
A>AG, G>GA, T>TC, C>CA, A>T) and one deletion
mutation (delA). Both heterozygous (n=35) and homozygous (n=1)
variant mutations were identified as demonstrated in Fig. 4. In total, 35 variant mutations
were previously not recorded in external databases and 30 of them
had their sequences of amino acids changed (Table II). The single homozygous
substitution variant (946A>T) accounts for the greatest
percentage (40%) among all other variants; however, among
heterozygous variants, two heterozygous variants (778A>AT and
793A>AT) constitute the second greatest percentage (33.30%)
among other mutation of VHL gene.
| Table IIVariants identified in VHL and KDR
genes in ccRCC patients analyzed with mutation DNA variant
analysis. |
Table II
Variants identified in VHL and KDR
genes in ccRCC patients analyzed with mutation DNA variant
analysis.
| | Chromosome
position | Mutation | Mutation
enotype |
Heterozygous/Homozygous | Variants | Variant percentage
(%) | Amino acid
change | External
database |
|---|
| VHL gene |
|---|
| 1 | 3:10183764 | Substitution | A>T | Homozygous | 946A>T | 40.00 |
Asparagine/Isoleucine | Not found |
| 2 | 3:10183623 | Substitution | A>AT | Heterozygous | 805A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 3 | 3:10183626 | Substitution | A>AT | Heterozygous | 808A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 4 | 3:10183638 | Substitution | A>AT | Heterozygous | 820A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 5 | 3:10183641 | Substitution | A>AT | Heterozygous | 823A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 6 | 3:10183656 | Substitution | A>AT | Heterozygous | 838A>AT | 30.00 | Glutamic
acid/Methionine | Not found |
| 7 | 3:10183671 | Substitution | A>AT | Heterozygous | 853A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 8 | 3:10183673 | Substitution | C>CA | Heterozygous | 855C>CA | 10.00 |
Lucine/Methionine | Not found |
| 9 | 3:10183686 | Substitution | A>AT | Heterozygous | 868A>AT | 30.00 | Glutamic
acid/Valine | Not found |
| 10 | 3:10183730 | Substitution | A>AT | Heterozygous | 912A>AT | 18.20 |
Asparagine/Tyrosine | Not found |
| 11 | 3:10183570 | Substitution | A>AT | Heterozygous | 752A>AT | 25 | none | Not found |
| 12 | 3:10183578 | Substitution | A>AT | Heterozygous | 760A>AT | 25 | Glutamic
acid/Valine | Not found |
| 13 | 3:10183581 | Substitution | A>AT | Heterozygous | 763A>AT | 25 | Glutamic
acid/Valine | Not found |
| 14 | 3:10183582 | Substitution | G>GA | Heterozygous | 764G>GA | 25 | Glutamic
acid/Valine | Not found |
| 15 | 3:10183596 | Substitution | A>AT | Heterozygous | 778A>AT | 33.30 | Glutamic
acid/Valine | Not found |
| 16 | 3:10183597 | Substitution | A>AT | Heterozygous | 793A>AT | 33.30 | Glutamic
acid/Valine | Not found |
| 17 | 3:10183612 | Substitution | A>AG | Heterozygous | 794A>AG | 22.20 | Glutamic
acid/Valine | Not found |
| 18 | 3:10183635 | Substitution | C>CT | Heterozygous | 817C>CT | 30.00 | Alanine/Valine | Not found |
| 19 | 3:10183655 | Substitution | G>GA | Heterozygous | 837G>GA | 10 | Glutamic
acid/Methionine | Not found |
| 20 | 3:10183611 | Substitution | A>AT | Heterozygous | 793A>AT | 33.30 | Glutamic
acid/Valine | Not found |
| 21 | 3:10183614 | Substitution | A>AT | Heterozygous | 796A>AT | 11.10 | Aspartic
acid/Valine | Not found |
| 22 | 3:10183764 | Deletion | A | | 946delA | 25 | none | Not found |
| 23 | 3:10183635 | Substitution | C>CT | Heterozygous | 817C>CT | 30.00 | Aspartic
acid/Valine | Not found |
| 24 | 3:10183579 | Substitution | G>GA | Heterozygous | 761G>GA | 12.50 | Glutamic
acid/Valine | Not found |
| 25 | 3:10183608 | Substitution | A>AT | Heterozygous | 790A>AT | 11.10 | Glutamic
acid/Valine | Not found |
| 26 | 3:10183624 | Substitution | G>GA | Heterozygous | 806G>GA | 10.00 | Glutamic
acid/Valine | Not found |
| 27 | 3:10183740 | Substitution | A>AT | Heterozygous | 922A>AT | 16.70 | Glutamic
acid/Valine | Not found |
| 28 | 3:10183672 | Substitution | A>AG | Heterozygous | 854A>AG | 9.10 | Glutamic
acid/Valine | Not found |
| 29 | 3:10183762 | Substitution | C>CT | Heterozygous | 944C>CT | 10 | none | Not found |
| 30 | 3:10183763 | Substitution | A>AG | Heterozygous | 945A>AG | 12.50 |
Isoleucine/Valine | Not found |
| 31 | 3:10183691 | Substitution | A>AT | Heterozygous | 873A>AT | 9.10 |
Methionine/Leucine | Not found |
| 32 | 3:10183753 | Substitution | C>CT | Heterozygous | 935C>CT | 8.30 | none | Not found |
| 33 | 3:10183759 | Substitution | C>CT | Heterozygous | 941C>CT | 9.10 | none | Not found |
| 34 | 3:10183756 | Substitution | C>CT | Heterozygous | 938C>CT | 8.30 | none | Not found |
| 35 | 3:10183719 | Substitution | T>TC | Heterozygous | 901T>TC | 9.10 |
Leucine/Proline | Not found |
| 36 | 3:10183764 | Substitution | A>AG | Heterozygous | 946A>AG | 12.50 |
Asparagine/serine | found |
| KDR gene |
| 1 | 4:55971029 | Substitution | C>CG | Heterozygous | 21234C>CG | 28.00 |
Proline/Alanine | Not found |
| 2 | 4:55970820 | Substitution | C>CT | Heterozygous | 21443C>CT | 8.30 | None | Not found |
| 3 | 4:55970985 | Substitution | G>GT | Heterozygous | 21278G>GT | 16.70 |
Lysin/Asparagine | Not found |
| 4 | 4:55970981 | Substitution | T>TC | Heterozygous | 21282T>TC | 12.50 |
Leucine/Proline | Not found |
| 5 | 4:55970980 | Substitution | T>TC | Heterozygous | 21283T>TC | 12.50 |
Leucine/Proline | Not found |
| 6 | 4:55970991 | Substitution | T>TC | Heterozygous | 21272T>TC | 16.70 | None | Not found |
| 7 | 4:55970969 | Substitution | T>TG | Heterozygous | 21294T>TG | 20 |
Tryptophane/Glycine | Not found |
| 8 | 4:55970819 | Substitution | A>AC | Heterozygous | 21444A>AC | 8.30 |
Threonine/Proline | Not found |
| 9 | 4:55971034 | Substitution | C>CT | Heterozygous | 21229C>CT | 25.30 |
Proline/Leucine | Not found |
| 10 | 4:55971000 | Substitution | C>CT | Heterozygous | 21263C>CT | 25.10 | None | Not found |
| 11 | 4:55970978 | Substitution | G>GC | Heterozygous | 21285G>GC | 12.50 | Aspartic
acid/Histidine | Not found |
| 12 | 4:55970970 | Substitution | T>TC | Heterozygous | 21293T>TC | 10.00 | None | Not found |
| 13 | 4:55970996 | Substitution | C>CG | Heterozygous | 21267C>CG | 25.00 | Proline/Aspartic
acid | Not found |
| 14 | 4:55970995 | Substitution | C>CA | Heterozygous | 21268C>CA | 25.00 | Proline/Aspartic
acid | Not found |
| 15 | 4:55970892 | Substitution | G>GA | Heterozygous | 21371G>GA | 7.10 | None | Not found |
| 16 | 4:55970925 | Substitution | T>TG | Heterozygous | 21338T>TG | 7.10 | Aspartic
acid/Histidine | Not found |
Regarding to KDR gene, 16 heterozygous substitution
variant mutations (C>CG, C>CT, G>GT, T>TC, A>AC,
G>GC, C>CA, G>GA) were recorded in exon 13; all of them
were not recorded previously in external databases. A total of 11
of these mutations are associated with changes in the sequence of
amino acids. Furthermore, the heterozygous variant (21234C>CG)
constitutes the largest percentage (28%) among other variants
(Table II).
Database mutations and gene
interactions
The GnomAD tool was used to retrieve the entire
recorded mutations of VHL and KDR genes in all types of cancer. VHL
had 386 mutations, while KDR had 1,595 mutations as presented in
Table SI.
Additional analysis of retrieved VHL and KDR gene
mutations in RCC was performed using Metagene (all genome-wide
studies in ICGC) and COSMIC databases as demonstrated in Tables SII and SIII. The number of mutations in VHL was
125 and 1,569 in the Metagene and COSMIC databases, respectively;
however, the number of mutations of the KDR gene was 229 and 27 in
the same two databases, respectively. In the Mutagen database the
most common type of mutation in the VHL gene of patients with RCC
was missense mutation followed by nonsense mutation. Out of 125
mutations, 66 were passengers followed by 34 drivers and 25
potential driver mutations. Regarding the KDR gene, the most common
type of mutation was missense followed by silent mutation, and
their status in cancer participation was mostly passenger (n=179)
followed by potential driver (n=34) and driver (n=16) mutations. In
the COSMIC database, the frameshift deletion (n=530) was the most
common type of mutation of VHL in patients with ccRCC followed by
missense-substitution (n=339), frameshift insertion (n=219) and
nonsense substitution (n=62). However, missense substitutions
(n=20) were common types of mutations of the KDR gene in patients
with ccRCC followed by frameshift deletions (n=3), nonsense
substitution (n=2) and substitution-coding silent (n=2).
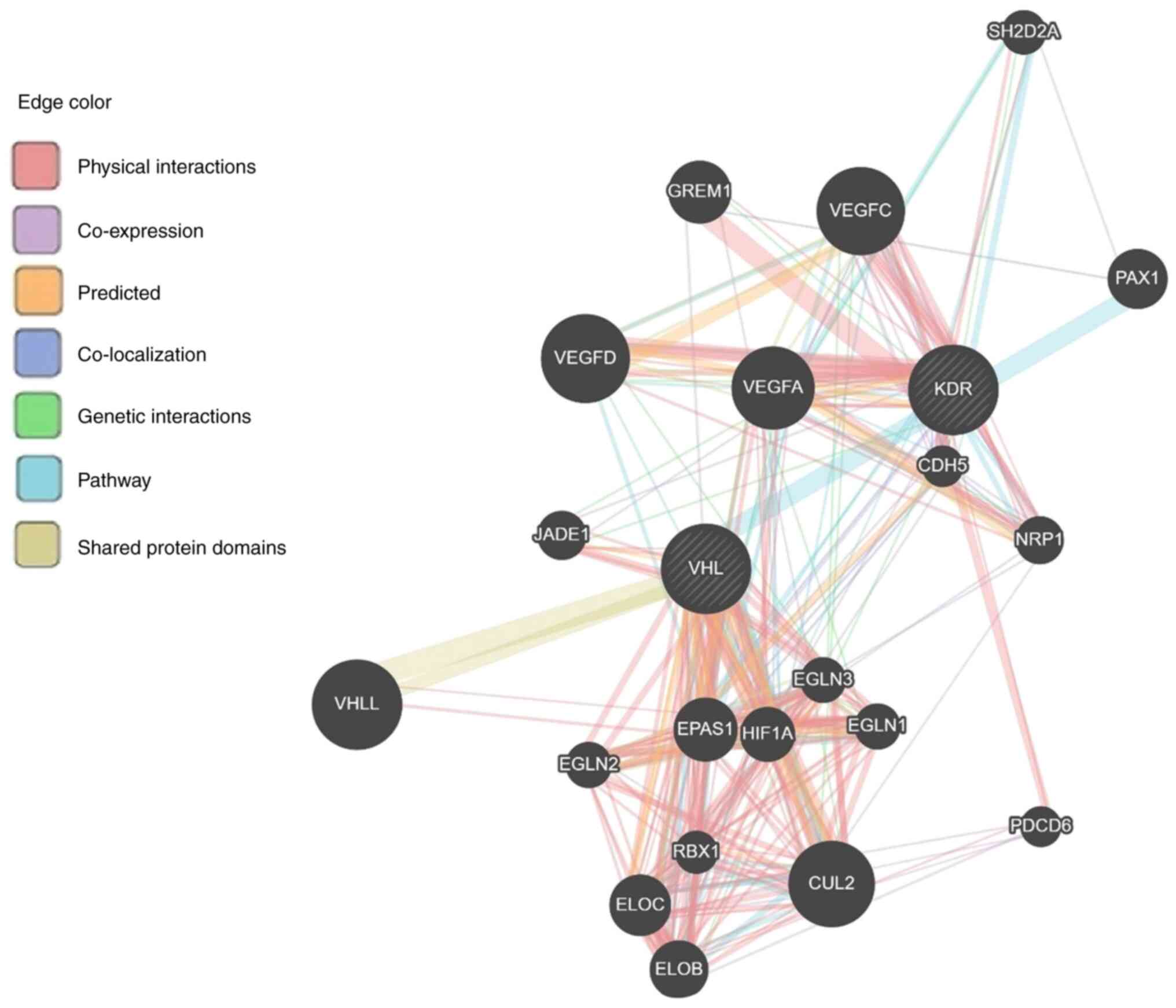
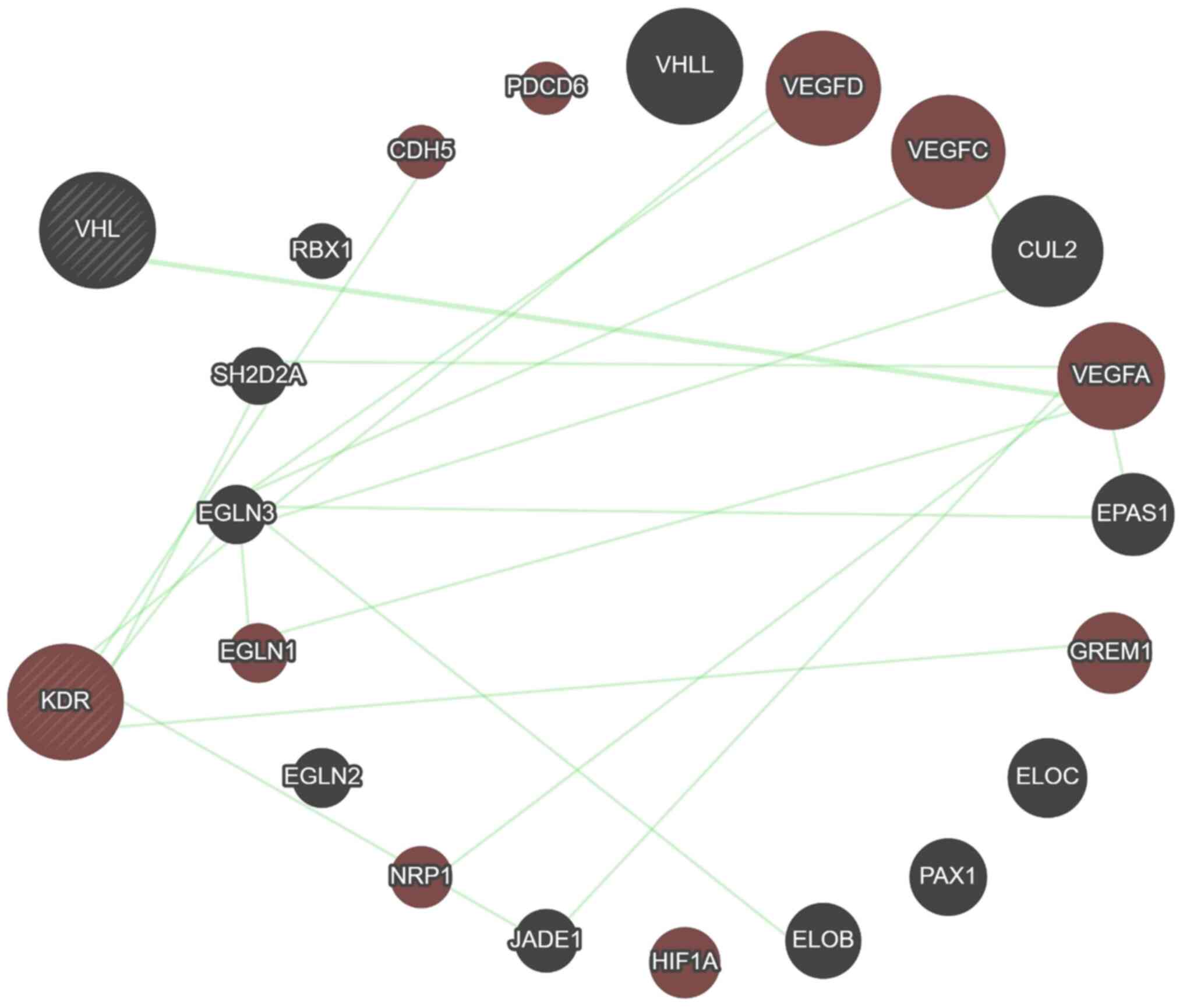
Moreover, data from the GeneMANIA predicting tool
revealed that 13 genes (RBX1, VHLL, CUL2, VEGFA, EPAS1, ELOC, PAX1,
ELOB, HIF1, JADE1, EGLN1, EGLN2 and EGLN3) and 12 genes (CDH5,
PDCD6, VEGFA, VEGFC, VEGFD, EPAS1, GREM1, HIF1, JADE1, NRP1, EGLN3
and SH2D2A) are associated with VHL and KDR genes, respectively,
mainly through physical interaction then co-expression, prediction,
colocalization, genetic interaction, pathway and minimum level by
sharing protein domain (Fig. 5 and
Table SIV). A total of five genes
(VEGFA, EPAS1, HIF1, JADE1 and EGLN3) are shared between VHL and
KDR genes, and this is considered a close relation between these
two genes. Furthermore, the KDR gene showed genetic interaction
with most of the genes having a role in angiogenesis (Fig. 6).
Discussion
In the present study, the sunitinib IC50
after 24 and 48 h was ~9x10-6 M, whereas after 72 h it
dropped to 5.26x10-6M. These results are very similar to
previous results demonstrating the cytotoxicity of sunitinib
against RCC cell lines. Sato et al (34) identified that the IC50
value of sunitinib against 786-O cells (3,000/well) was 5 µM after
a 48-h incubation. This disparity in the results might be
attributed to the initial cell count used in the experiment as the
current study involved plating 4,000 cells in each well of a
96-well plate, which could have affected the observed
IC50 value due to variations in cell density. Another
study reported that sunitinib IC50 was 3.99 µM against
Caki-1 cells after 48 h (35). In
this case, the discrepancy with the present results might be
attributed to the differences in the cell line.
In the current study, ccRCC 786-O cells were exposed
for 24, 48, 72, and 96 h to several concentrations of BAY-876. The
results demonstrated that BAY-876 can reduce 786-O cells in a
dose-dependent manner and the IC50 was
53.56x10-6 M. As RCC specimens demonstrated a remarkably
significant rise in the expression levels of GLUT1 compared with
the corresponding normal kidney tissue (36,37),
the present finding proves that reducing cell glucose intake by
inhibiting GLUT1 through BAY-876 treatment, could represent a new
strategy to suppress the growth of ccRCC. Moreover, the present
result is consistent with a recent study (38) showing that BAY-876 has an
inhibitory effect against bladder urothelial carcinoma (BLCA) cell
lines and tumor inhibition effect in xenograft models. It was
proposed that CLUTI inhibition through BAY-876 could be an
effective approach to inhibiting the growth of BLCA tumors.
Furthermore, another study (17)
revealed that BAY-876 is a strong blocker of GLUT1 activity,
mechanisms of glycolysis and ovarian cancer cell growth in ovarian
cancer cell lines, as well as cell line-derived xerographs and
ovarian cancer patient-derived xenografts.
The present results identified that the combination
of BAY-876 and sunitinib has a significant cytotoxic effect on
786-O cells at shorter incubation times (after 24 and 48 h)
compared with each drug alone, which had no cybotactic effect at
these intervals. This combination may eliminate the cell by
reducing the sugar intake by blocking GLUT1 transporters as well as
VEGF, PDGF and c-Kit receptors (39). The multi-shunt elimination of 786-O
cells during drug combination exposure may be attributable to a
reduction in the duration of cell death when compared with each
drug administered separately. This could be a promising advancement
in the treatment of RCC.
RCC presents a contradictory aspect of the action of
endogenous H2S. Numerous studies demonstrated an
increase in H2S levels or H2S-producing
enzymes in RCC cell lines, xenograft models and ccRCC tissues.
These findings suggested a correlation between H2S and
RCC growth and progression (40,41).
On the contrary, another study argued that the expression of
H2S-producing enzymes in ccRCC was reduced (42). The findings of the present study
indicated that the use of NaSH alone resulted in a significant
increase in cell death compared with the cells that were not
treated. This finding aligns with the results of a previous study
which demonstrated that NaSH has a significant impact on reducing
cell viability in three different breast cancer cell lines (MCF-10,
MCF-7 and MDA-MB-23) (43). The
aforementioned study also demonstrated that exogenous
H2S can suppress the progression of RCC by inhibiting
the P13K/AKT pathway as well as by inducing apoptosis.
The cytotoxic activity of both sunitinib and BAY-876
increased when combined with NaSH. Overall, the combination of NaSH
and BAY-876 could represent a novel approach to treating resistant
metastatic ccRCC. If they are used in treatment, they may reduce
the serious health impact and adverse reaction of sunitinib and
other TKIs.
The clinicopathological characteristics of the
patients in the present study revealed a slightly higher incidence
rate of ccRCC in male patients (56.6%) compared with female
patients (44.4%). However, a recent study demonstrated a
significantly higher incidence rate of ccRCC in male patients (80%)
compared with female patients (20%) (44). In addition, the TNM staging
revealed that the incidence rates for stages I, II and III were
76.66, 3.33 and 20%, respectively.
Inactivation of the gene encoding the tumor
suppressor VHL mutations is the most prevalent in ccRCC (45,46).
This present study revealed that the incidence of intragenic VHL
mutation was 43.7%, which is marginally lower than the percentage
observed in a study conducted in Japan, which was 51% (47). This may be because only exon 1 was
sequenced in the present research, whereas the entire gene was
sequenced in the study by Kondo et al (47). In addition, the present study
identified 36 missense SNP mutations in exon 1 of the VHL gene.
This funding is consistent with the findings of a recent study
which demonstrated that all mutations in VHL are missense mutations
in ccRCC (48), and with COSMIC
databases, which demonstrated that missense mutation is superior to
other types of mutations in RCC. Based on this number of point
mutations in VHL, mostly with an amino acid change (30 out of 36),
the present study suggested that mutations in this gene could be
the primary cause of ccRCC and confirmed that TKIs such as
sunitinib may be one of the best options for inhibiting the growth
of this type of kidney cancer, as they inhibit angiogenesis by
inhibiting the downstream pathways of VHL/HIF.
VEGFR-2 serves a crucial role as a primary mediator
in tumor angiogenesis and is regarded as a significant target for
therapeutic interventions to counteract angiogenesis. Several
anti-angiogenic medications, including ramucirumab, sunitinib,
axitinib and sorafenib, have been developed to target this pathway
(49). A previous study revealed
that the rs1870377 A>T genetic polymorphism of VEGFR-2 is
associated with the prognosis of gastric cancer (50). The present study demonstrated that
there were 16 heterozygous missense variants in exon 13 of KDR,
which is consistent with data from both the COSMIC and mutagen
databases. This may explain the function of KDR gene mutation in
the development of ccRCC and resistance to numerous anti-angiogenic
drugs. KDR mutation testing is crucial for guiding the choice of
therapy and improving patient outcomes.
Moreover, the present study found that BAY-876 and
NaSH could improve sunitinib's anticancer efficacy in ccRCC cell
lines, suggesting a novel treatment approach. It also found that
VHL mutation is the main cause of ccRCC development and KDR
mutation is necessary for angiogenesis. Testing these mutations
might help choose the right therapy and understand the illness
process. To the best of the authors' knowledge, the present study
is the first to identify a significant number of VHL and KDR gene
mutations in patients with ccRCC. By interpreting these gene
mutations and relating their detection with relevant
mutation-related datasets, the molecular mechanisms that initiate
and progress ccRCC were elucidated. The present investigation is
important in the context of RCC research and lays the groundwork
for future studies. However, additional research is needed to
confirm the current outcomes on other cell lines, also to determine
the cellular and physiological effects of these agents. Functional
studies could investigate the molecular effects of these mutations,
their effects on clinical outcomes in larger groups, their
predictive value for treatment response and genomic analysis to
identify more mutations and variations. Notably, the significance
of tracking response rate and cancer regression rate over time is
recognized as a crucial avenue for future research and it is
intended by the authors to contemplate integrating this data into
subsequent research endeavors or offering a more comprehensive
analysis in subsequent studies.
Supplementary Material
Agarose gel electrophoresis of PCR
product for tumor suppressor gene Von Hippel Lindau and kinase
insert domain receptor genes. (A) Represent amplification of exon I
of VHL gene. (B) Represent the amplification segment of axon 13 of
KRD gene. Lane 1: Ladder. Lane 2-16 demonstrate PCR products of
ccRCC patients. The bands for both VHL and KRD fragments for all
patients were located within the expected fragment size (VHL=260 bp
and KDR=290 bp).
Summary of mutations in the VHL and
KDR genes in all types of cancer retrieved from the gnomAD
database.
Summary of mutations in the VHL and
KDR genes in renal cell carcinoma retrieved from the Mutagene
database.
Summary of mutations in the VHL and
KDR genes in ccRCC retrieved from the COSMIC database.
The interaction between VHL ad KDR
gene and their related genes retrieved from GeneMania database
Acknowledgements
The authors would like to thank Dr Abbas Salihi from
Salahaddin University in Erbil, Salahaddin Universality Research
Center as well as the Histopathology department and laboratory
staff members at Nanakaly Hospital, Rizgary Hospital, and Par
Hospital in Erbil, Iraq, for providing samples.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PSH conceived and designed the experiments,
performed the experiments, analyzed the data, prepared figures
and/or tables, authored or reviewed drafts of the article. KAM
conceived and designed the experiments, performed the experiments,
authored, or reviewed drafts of the article. PSH and KAM confirm
the authenticity of all the raw data. Both authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no. 45/90)
by the Human Research Ethics Committee of Affiliated Salahaddin
University (Erbil, Iraq). Written informed consent forms were
obtained from the participants approving the use of their tissues
for investigation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bahadoram S, Davoodi M, Hassanzadeh S,
Bahadoram M, Barahman M and Mafakher L: Renal cell carcinoma: An
overview of the epidemiology, diagnosis, and treatment. G Ital
Nefrol. 39:32–47. 2022.PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Monjaras-Avila CU, Lorenzo-Leal AC,
Luque-Badillo AC, D'Costa N, Chavez-Muñoz C and Bach H: The tumor
immune microenvironment in clear cell renal cell carcinoma. Int J
Mol Sci. 24(7946)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kase AM, George DJ and Ramalingam S: Clear
cell renal cell carcinoma: From biology to treatment. Cancers
(Basel). 15(665)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Padala SA and Barsouk A, Thandra KC,
Saginala K, Mohammed A, Vakiti A, Rawla P and Barsouk A:
Epidemiology of renal cell carcinoma. World J Oncol. 11:79–87.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jonasch E, Walker CL and Rathmell WK:
Clear cell renal cell carcinoma ontogeny and mechanisms of
lethality. Nat Rev Nephrol. 17:245–261. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Nickerson ML, Jaeger E, Shi Y, Durocher
JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko
V, et al: Improved identification of von Hippel-Lindau gene
alterations in clear cell renal tumors. Clin Cancer Res.
14:4726–4734. 2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dizman N, Philip EJ and Pal SK: Genomic
profiling in renal cell carcinoma. Nat Rev Nephrol. 16:435–451.
2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Williamson SR: Clear cell papillary renal
cell carcinoma: An update after 15 years. Pathology. 53:109–119.
2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sharma R, Kadife E, Myers M, Kannourakis
G, Prithviraj P and Ahmed N: Determinants of resistance to VEGF-TKI
and immune checkpoint inhibitors in metastatic renal cell
carcinoma. J Exp Clin Cancer Res. 40(186)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3(17009)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Makino T, Kadomoto S, Izumi K and Mizokami
A: Epidemiology and prevention of renal cell carcinoma. Cancers
(Basel). 14(4059)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Choi WSW, Boland J and Lin J:
Hypoxia-inducible factor-2α as a novel target in renal cell
carcinoma. J Kidney Cancer VHL. 8:1–7. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik
C, Kim ST, et al: Sunitinib versus interferon alfa in metastatic
renal-cell carcinoma. N Engl J Med. 356:115–124. 2007.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jin J, Xie Y, Zhang JS, Wang JQ, Dai SJ,
He WF, Li SY, Ashby CR Jr, Chen ZS and He Q: Sunitinib resistance
in renal cell carcinoma: From molecular mechanisms to predictive
biomarkers. Drug Resist Updat. 67(100929)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rini BI, Tamaskar I, Shaheen P, Salas R,
Garcia J, Wood L, Reddy S, Dreicer R and Bukowski RM:
Hypothyroidism in patients with metastatic renal cell carcinoma
treated with sunitinib. J Natl Cancer Inst. 99:81–83.
2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ma Y, Wang W, Idowu MO, Oh U, Wang XY,
Temkin SM and Fang X: Ovarian cancer relies on glucose transporter
1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876.
Cancers (Basel). 11(33)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mantle D and Yang G: Hydrogen sulfide and
metal interaction: The pathophysiological implications. Mol Cell
Biochem. 477:2235–2248. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zaorska E, Tomasova L, Koszelewski D,
Ostaszewski R and Ufnal M: hydrogen sulfide in pharmacotherapy,
beyond the hydrogen sulfide-donors. Biomolecules.
10(323)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Khattak S, Rauf MA, Khan NH, Zhang QQ,
Chen HJ, Muhammad P, Ansari MA, Alomary MN, Jahangir M, Zhang CY,
et al: Hydrogen sulfide biology and its role in cancer. Molecules.
27(3389)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M,
Moore PK and Bian JS: A review of hydrogen sulfide synthesis,
metabolism, and measurement: Is modulation of hydrogen sulfide a
novel therapeutic for cancer? Antioxid Redox Signal. 31:1–38.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Powell CR, Dillon KM and Matson JB: A
review of hydrogen sulfide (H(2)S) donors: Chemistry and potential
therapeutic applications. Biochem Pharmacol. 149:110–123.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen G, Zhang Y and Wu X: 786-0 renal
cancer cell line-derived exosomes promote 786-0 cell migration and
invasion in vitro. Oncol Lett. 7:1576–1580. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gu L, Sang Y, Nan X, Zheng Y, Liu F, Meng
L, Sang M and Shan B: circCYP24A1 facilitates esophageal squamous
cell carcinoma progression through binding PKM2 to regulate
NF-κB-induced CCL5 secretion. Mol Cancer. 21(217)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Warren AY and Harrison D: WHO/ISUP
classification, grading and pathological staging of renal cell
carcinoma: Standards and controversies. World J Urol. 36:1913–1926.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Elkassem AA, Allen BC, Sharbidre KG,
Rais-Bahrami S and Smith AD: Update on the role of imaging in
clinical staging and restaging of renal cell carcinoma based on the
AJCC 8th edition, from the AJR special series on cancer staging.
AJR Am J Roentgenol. 217:541–555. 2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Delahunt B, Eble JN, Samaratunga H,
Thunders M, Yaxley JW and Egevad L: Staging of renal cell
carcinoma: Current progress and potential advances. Pathology.
53:120–128. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Abid MN, Qadir FA and Salihi A:
Association between the serum concentrations and mutational status
of IL-8, IL-27 and VEGF and the expression levels of the hERG
potassium channel gene in patients with colorectal cancer. Oncol
Lett. 22(665)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Alves MR, Carneiro FC, Lavorato-Rocha AM,
da Costa WH, da Cunha IW, de Cássio Zequi S, Guimaraes GC, Soares
FA, Carraro DM and Rocha RM: Mutational status of VHL gene and its
clinical importance in renal clear cell carcinoma. Virchows Arch.
465:321–330. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Housein Z, Kareem TS and Salihi A: In
vitro anticancer activity of hydrogen sulfide and nitric oxide
alongside nickel nanoparticle and novel mutations in their genes in
CRC patients. Sci Rep. 11(2536)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gudmundsson S, Singer-Berk M, Watts NA,
Phu W, Goodrich JK and Solomonson M: Genome Aggregation Database
Consortium. Rehm HL, MacArthur DG and O'Donnell-Luria A: Variant
interpretation using population databases: Lessons from gnomAD. Hum
Mutat. 43:1012–1030. 2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sondka Z, Bamford S, Cole CG, Ward SA,
Dunham I and Forbes SA: The COSMIC cancer gene census: Describing
genetic dysfunction across all human cancers. Nat Rev Cancer.
18:696–705. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Franz M, Rodriguez H, Lopes C, Zuberi K,
Montojo J, Bader GD and Morris Q: GeneMANIA update 2018. Nucleic
Acids Res. 46:W60–W64. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sato H, Siddig S, Uzu M, Suzuki S, Nomura
Y, Kashiba T, Gushimiyagi K, Sekine Y, Uehara T, Arano Y, et al:
Elacridar enhances the cytotoxic effects of sunitinib and prevents
multidrug resistance in renal carcinoma cells. Eur J Pharmacol.
746:258–266. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Amaro F, Pisoeiro C, Valente MJ, Bastos
ML, de Pinho PG, Carvalho M and Pinto J: Sunitinib versus pazopanib
dilemma in renal cell carcinoma: New Insights into the in vitro
metabolic impact, efficacy, and safety. Int J Mol Sci.
23(9898)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Singer K, Kastenberger M, Gottfried E,
Hammerschmied CG, Büttner M, Aigner M, Seliger B, Walter B,
Schlösser H, Hartmann A, et al: Warburg phenotype in renal cell
carcinoma: high expression of glucose-transporter 1 (GLUT-1)
correlates with low CD8(+) T-cell infiltration in the tumor. Int J
Cancer. 128:2085–2095. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Abdolahi M, Alam M, Ghaffarpasand A, Nouri
F, Badkoobeh A, Golkar M, Abassi K and Torbati P: Assessment of the
expression of GLUT1 in renal cell carcinoma and its various
subtypes. Open Access Maced J Med Sci. 10:2581–2585. 2022.
|
|
38
|
Wang X, He H, Rui W, Zhang N, Zhu Y and
Xie X: TRIM38 triggers the uniquitination and degradation of
glucose transporter type 1 (GLUT1) to restrict tumor progression in
bladder cancer. J Transl Med. 19(508)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kollmannsberger C, Soulieres D, Wong R,
Scalera A, Gaspo R and Bjarnason G: Sunitinib therapy for
metastatic renal cell carcinoma: recommendations for management of
side effects. Can Urol Assoc J. 1:S41–S54. 2007.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Sonke E, Verrydt M, Postenka CO, Pardhan
S, Willie CJ, Mazzola CR, Hammers MD, Pluth MD, Lobb I, Power N, et
al: Inhibition of endogenous hydrogen sulfide production in
clear-cell renal cell carcinoma cell lines and xenografts restricts
their growth, survival and angiogenic potential. Nitric Oxide.
49:26–39. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shackelford RE, Abdulsattar J, Wei EX,
Cotelingam J, Coppola D and Herrera GA: Increased nicotinamide
phosphoribosyltransferase and cystathionine-β-synthase in renal
oncocytomas, renal urothelial carcinoma, and renal clear cell
carcinoma. Anticancer Res. 37:3423–3427. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Breza J Jr, Soltysova A, Hudecova S,
Penesova A, Szadvari I, Babula P, Chovancova B, Lencesova L, Pos O,
Breza J, et al: Endogenous H(2)S producing enzymes are involved in
apoptosis induction in clear cell renal cell carcinoma. BMC Cancer.
18(591)2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Dong Q, Yang B, Han JG, Zhang MM, Liu W,
Zhang X, Yu HL, Liu ZG, Zhang SH, Li T, et al: A novel hydrogen
sulfide-releasing donor, HA-ADT, suppresses the growth of human
breast cancer cells through inhibiting the PI3K/AKT/mTOR and
Ras/Raf/MEK/ERK signaling pathways. Cancer Lett. 455:60–72.
2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sekar H, Krishnamoorthy S, Kumaresan N,
Chandrasekaran D, Ramaswamy P, Sundaram S and Raj N:
Clinicopathological comparison of VHL expression as a prognostic
tumor marker in renal cell carcinoma: A single center experience.
Niger J Clin Pract. 24:614–620. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Audenet F, Yates DR, Cancel-Tassin G,
Cussenot O and Rouprêt M: Genetic pathways involved in
carcinogenesis of clear cell renal cell carcinoma: Genomics towards
personalized medicine. BJU Int. 109:1864–1870. 2012.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Maxwell PH, Wiesener MS, Chang GW,
Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER and
Ratcliffe PJ: The tumour suppressor protein VHL targets
hypoxia-inducible factors for oxygen-dependent proteolysis. Nature.
399:271–275. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
47
|
Kondo K, Yao M, Yoshida M, Kishida T,
Shuin T, Miura T, Moriyama M, Kobayashi K, Sakai N, Kaneko S, et
al: Comprehensive mutational analysis of the VHL gene in sporadic
renal cell carcinoma: Relationship to clinicopathological
parameters. Genes Chromosomes Cancer. 34:58–68. 2002.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin PH, Huang CY, Yu KJ, Kan HC, Liu CY,
Chuang CK, Lu YC, Chang YH, Shao IH and Pang ST: Genomic
characterization of clear cell renal cell carcinoma using targeted
gene sequencing. Oncol Lett. 21(169)2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Shah AA, Kamal MA and Akhtar S: Tumor
angiogenesis and VEGFR-2: Mechanism, pathways and current
biological therapeutic interventions. Curr Drug Metab. 22:50–59.
2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhu X, Wang Y, Xue W, Wang R, Wang L, Zhu
ML and Zheng L: The VEGFR-2 protein and the VEGFR-2 rs1870377
A>T genetic polymorphism are prognostic factors for gastric
cancer. Cancer Biol Ther. 20:497–504. 2019.PubMed/NCBI View Article : Google Scholar
|