Introduction
Acetaminophen (APAP), also known as paracetamol, is
a safe analgesic and antipyretic agent at therapeutic dose
(1). It has been widely applied in
the clinic (2–4). In general, an overdose of APAP of
10–15 g can cause serious toxicity and is harmful to the liver and
the kidneys (5,6). APAP is easily available and cheap,
and thus patients may easily receive an overdose. This is one of
the reasons that APAP constitutes the most common cause of
self-poisoning in numerous countries (7–9). In
order to study APAP overdose-induced liver and acute kidney damage,
a number of animal and cell models have been established. Studies
in these models have shown that treatment with high doses of APAP
(300–2,500 mg/kg) can cause hepatotoxicity and nephrotoxicity in
vivo (10–14), and doses >0.005 mol/l can induce
cytotoxicity on kidney and liver cells (15–20).
Previous studies have shown that APAP can induce apoptosis or
necrosis on different cell models (14,19,21),
and that high-dose APAP treatment can increase oxidative stress,
decrease the glutathione level and activate MAPK signaling
pathways, resulting in cell cytotoxicity (14,16,20,22–25).
A number of recent studies have indicated that
high-dose APAP treatment causes liver and kidney failure (26–28).
However, other studies reported that high-dose APAP treatment also
exerts anticancer effects. These studies showed that APAP can
induce cytotoxicity on neuroblastoma (SH-SY5Y cells), hepatoma
(HuH7 cells) and breast cancer (FM3A cells) (29–33).
These studies also demonstrated, in different tumor cell types,
that APAP-induced cell death is related to the proteins NF-κB,
members of the Bcl-2 family, and the glycogen synthase kinase-3. In
addition, APAP can enhance the chemotherapeutic anticancer effects
of drugs used to treat neuroblastoma, leukemia and ovarian
carcinoma (30,34,35).
According to the above studies, APAP can activate different
cytotoxic mechanisms in liver, kidney and tumor cells (14,19,21,31,36).
To date, most studies have focused on the mechanisms of
APAP-induced cytotoxicity and on how to prevent high-dose
APAP-related poisoning of the liver and the kidneys. However,
whether APAP can enhance cell proliferation remains unclear.
Kidney tubular epithelial cell damage can induce
renal failure (37–40). Kidney fibrosis, via fibroblast
proliferation, can also cause renal failure (41–43).
Therefore, both kidney tubular cell damage and fibroblast
proliferation can cause kidney dysfunction. Recently, high-dose
APAP-induced nephrotoxicity was reported and investigated (13,22,44–47).
These studies found that high-dose APAP treatment can induce kidney
tubular cell death in animal and cell models. In addition, numerous
studies have demonstrated that high-dose APAP treatment can induce
an increase in oxidative stress, causing tubular cell death through
necrosis or the apoptotic pathway (13,22,44,47,48).
However, there is no evidence that APAP can cause kidney
dysfunction by inducing fibroblast proliferation. The present study
is the first to demonstrate, to the best of our knowledge, that
high doses of APAP (7.94 mM) can inhibit cell survival in kidney
tubular cells (NRK-52E), while promoting cell proliferation in
kidney interstitial fibroblasts (NRK-49F).
In addition, APAP can induce different cytotoxic
mechanisms on different hepatoma cell lines. APAP can induce
caspase-dependent apoptosis on hepatoma HuH7 and SK-Hep1 cells
(31,49) and induces apoptosis and necrosis on
hepatoma HepG2 cells (50).
Additionally, a study demonstrated that high-dose APAP treatment
can inhibit DOX-induced cell death in hepatoma HepG2 cells
(36). Although APAP-induced
apoptosis of hepatoma Hep3B cells was reported (51), the underlying mechanisms are still
unclear.
Materials and methods
Materials
Luminol, lucigenin and Hoechst 33342 were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Transforming growth factor
(TGF)-β was purchased from R&D Systems (Minneapolis, MN, USA).
The MTT assay kit was purchased from Bio Basic Canada, Inc.
(Markham, ON, Canada). The caspase-9 substrate
acetyl-Leu-Glu-His-Asp-p-nitroanilide (Ac-LEHD-pNA), the
caspase-3-like substrate
acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNA) and the
caspase-8 substrate acetyl-Ile-Glu-Thr-Asp-p-nitroanilide
(Ac-IETD-pNA) were purchased from AnaSpec, Inc. (San Jose, CA,
USA). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium
(DMEM), non-essential amino acids, L-glutamine and
penicillin/streptomycin were purchased from Gibco-BRL (Carlsbad,
CA, USA).
Cell lines and cultures
The rat kidney cell lines NRK-52E (tubular
epithelial cells) and NRK-49F (fibroblasts) and Hep3B cells were
purchased from the Bioresource Collection and Research Center
(Hsinchu, Taiwan). These cell lines were cultured in DMEM medium
supplemented with 10% FBS, 2 mM L-glutamine, 100 IU/ml
penicillin/streptomycin and 0.1 mM non-essential amino acids, and
were maintained at 37°C in a humidified atmosphere containing 5%
CO2, as in (52,53).
Cell survival assay
Survival rates of NRK-52E, NRK-49F and Hep3B cells
were determined with the MTT assay as previously described
(54,55). Briefly, cells were cultured in
96-well plates. On the second day, cells were divided into the
control and experimental groups. After cells were treated with 7.94
nM APAP, 0.794 nM APAP, 0.0794 nM APAP and 1nM TGF-B, respectively,
cell survival rates were measured every day. The MTT assay was
conducted daily according to the manufacturer’s instructions.
Absorbance was measured at 570 nm using a multi-well ELISA reader
(Molecular Devices, Sunnyvale, CA, USA).
Quantification of
H2O2 and O2−
levels
H2O2 and
O2− levels were measured using a
lucigenin-amplified chemiluminescence method, as in (56,57).
Briefly, 200 μl of cell lysate was mixed with 0.2 mmol/l of luminol
solution (100 μl) for the quantification of the
H2O2 level, or with 0.1 mmol/l of lucigenin
solution (500 μl) for the quantification of the
O2− level. Measurements were then performed
on the CLA-FSI chemiluminescence analyzing system (Tohoku
Electronic Industrial Co., Ltd., Sendal, Japan). Each assay was
performed four times and results were expressed as the
chemiluminescence count per 10 sec.
Nuclear observation
Nuclear morphology was observed by nuclear staining
with Hoechst 33342. Cells were treated with Hoechst 33342 (10
μg/ml) for 10 min. Nuclear condensation and DNA fragmentation were
observed under a fluorescence microscope (excitation, 352;
emission, 450 nm; Olympus BX61; Olympus Corporation, Tokyo, Japan),
as described in previous studies (58,59).
Caspase activity assay
Cells were treated with lysis buffer (50 mM
Tris-HCl, 120 mM NaCl, 1 mM EDTA, 1% NP-40, pH 7.5), and then 1 μM
protease inhibitors (Cocktail set 539131; Merck KGaA, Darmstadt,
Germany) were added. Cell pellets were obtained by centrifugation
(15,000 × g, 4°C, 30 min). Caspase-3, -8 and -9 activities were
determined based on assays described in previous studies (60–62).
Briefly, 40 μl of cell lysate (80 μg total protein) were mixed with
158 μl reaction buffer (20% glycerol, 0.5 mM EDTA, 5 mM
dithiothreitol, 100 mM HEPES, pH 7.5) and 2 μl fluorogenic
substrate (Ac-LEHD-pNA, Ac-DEVD-pNA, or Ac-IETD-pNA) and were
incubated at 37°C for 6 h. The absorbance of the cleaved
fluorogenic substrate was detected at 405 nm (A405) in a FLx800™
fluorescence microplate reader (BioTek Instruments, Inc., Winooski,
VT, USA). The fold increase (FI) in caspase activity was calculated
using the following formula: FI = (A405sample −
A405control)/A405control.
Data analysis
Data were obtained from four independent triplicate
experiments and are presented as mean values of all data, with
related standard deviations (SD).
Results
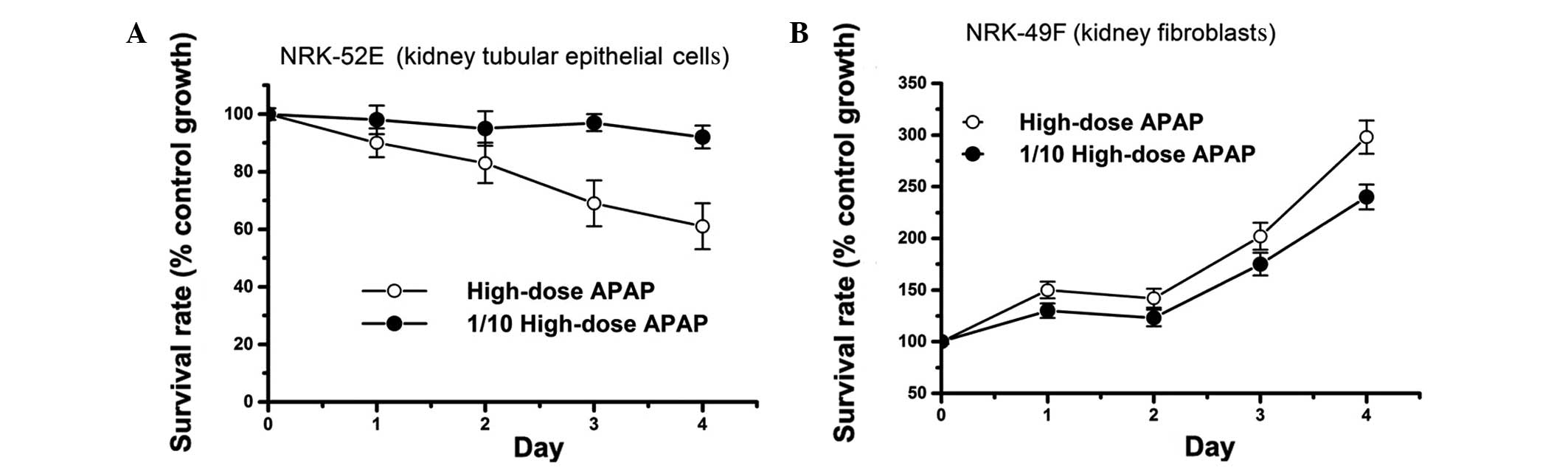
APAP treatment reduces the survival rate
of kidney tubular epithelial cells, while inducing proliferation of
kidney fibroblasts
Previous studies showed that a high dose of APAP
(>5 mM) can cause cell cytotoxicity in vitro (15–20).
In accordance with these studies, we also found that high-dose
(7.94 mM) APAP treatment reduces the survival rate of kidney
tubular epithelial cells (NRK-52E line), in a time-dependent manner
(Fig. 1A). The survival rate of
NRK-52E cells did not decrease upon treatment with 1/10 of the high
dose of APAP compared to high-dose treatment (Fig. 1A). These results suggest that
APAP-induced cell cytotoxicity is dependent on APAP concentration
and incubation time. However, to our surprise, although high-dose
APAP treatment decreased the survival rate of NRK-52E cells, it
promoted cell proliferation of kidney fibroblasts (NRK-49F line)
(Fig. 1B). This was also observed
upon treatment with 1/10 of the high dose of APAP (Fig. 1B). It is well established in the
clinic that both tubular epithelial cell damage and kidney fibrosis
can induce renal failure (13,22,41,43,47,48,63,64).
Therefore, our findings indicate that APAP-induced renal failure
may not only relate to the inhibition of tubular epithelial cell
survival, but also to the promotion of renal fibroblast
proliferation.
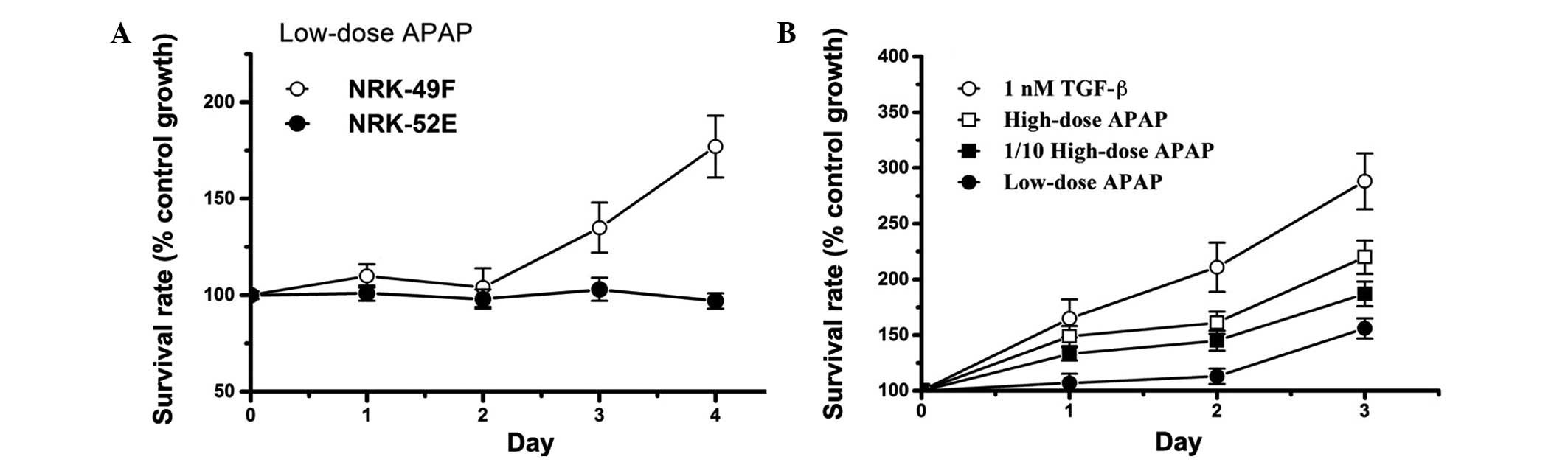
Low-dose APAP treatment induces
proliferation of kidney fibroblasts
Previous studies have demonstrated that high-dose
APAP treatment can inhibit tubular epithelial cell survival to
induce renal failure (13,22,43,47,48).
In the present study, as shown in Fig.
1, high-dose APAP treatment inhibited growth of tubular
epithelial cells, and induced proliferation of kidney fibroblasts.
In patients with kidney fibrosis, it is important to prevent
fibroblast proliferation, which further aggravates their condition.
In order to enhance our understanding on the effects of APAP
treatment on patients with fibrosis, it is therefore valuable to
investigate whether low doses of APAP (below the therapeutic dose)
can induce fibroblast proliferation. In this study, low-dose APAP
treatment was applied on kidney fibroblasts to study its effects on
cell growth. It is notable that low-dose APAP treatment did not
inhibit cell survival of NRK-52E cells, while low-dose treatment
induced cell proliferation in the fibroblast cell line NRK-49F
(Fig. 2A). In addition, APAP
induced fibroblast proliferation similarly to the treatment with
the positive control TGF-β, and in a dose-dependent manner
(Fig. 2B). APAP has not been
reported to be toxic to liver and kidney cells at doses below the
therapeutic dose in the clinic. However in our experiments, a low
dose of APAP induced fibroblast proliferation, which may have
harmful effects in patients with fibrosis. Thus, our study suggests
that these patients may be sensitive to even low doses of APAP.
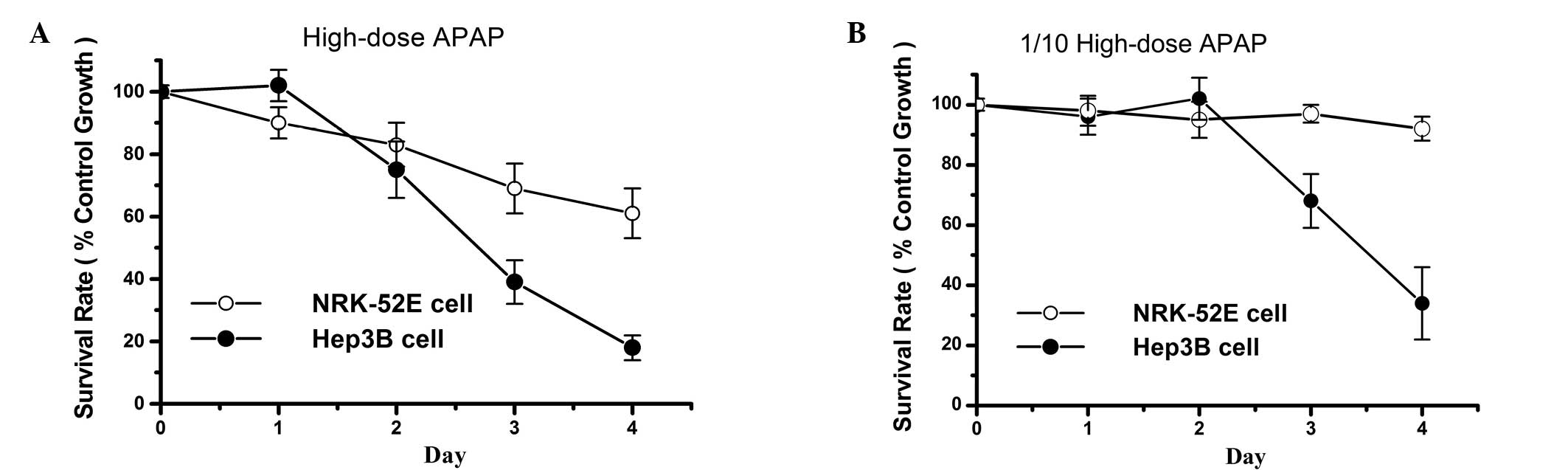
The cytotoxic effects of APAP are more
prominent in Hep3B compared to NRK52 cells
High-dose APAP treatment induced cytotoxic effects
not only in the tubular cell line NRK-52E, but also in the hepatoma
cell line Hep3B (Fig. 3A). Cell
survival rates of treated Hep3B cells were lower compared to those
observed in NRK-52E cells. However, at an APAP concentration that
was 1/10 of the high dose (therapeutic dose), no obvious cytotoxic
effects were observed in NRK-52E cells, while the survival rate of
Hep3B cells was markedly reduced (Fig.
3B). Therefore, APAP exerts more prominent cytotoxic effects on
Hep3B compared to NRK-52E cells. These results indicate that APAP,
at a non-toxic concentration for healthy tubular cells, may exert
an antitumor effect on hepatoma cells.
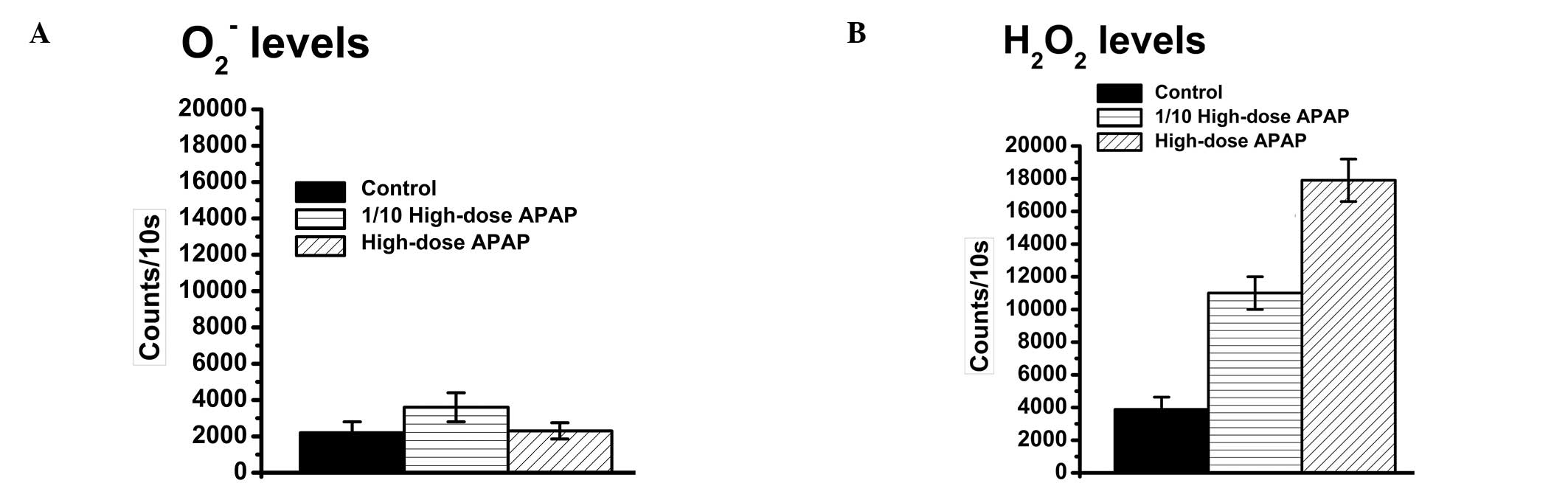
APAP treatment increases apoptosis of
Hep3B cells via an increase in the H2O2
level
APAP-induced cytotoxic effects that relate to an
increase in the generation of reactive oxygen species (ROS) were
previously reported (65,66). However, it is still unclear which
ROS elements are increased upon APAP treatment.
O2− and H2O2 are two
commonly found ROS types in the cells. O2−
and H2O2 levels were thus quantified
following APAP treatment. The result showed that APAP causes an
increase in the H2O2 (Fig. 4A), but not in the
O2−, level in Hep3B cells (Fig. 4B). Therefore, APAP-induced
cytotoxicity is possibly related to H2O2 but
not to O2−. In addition, microscopic
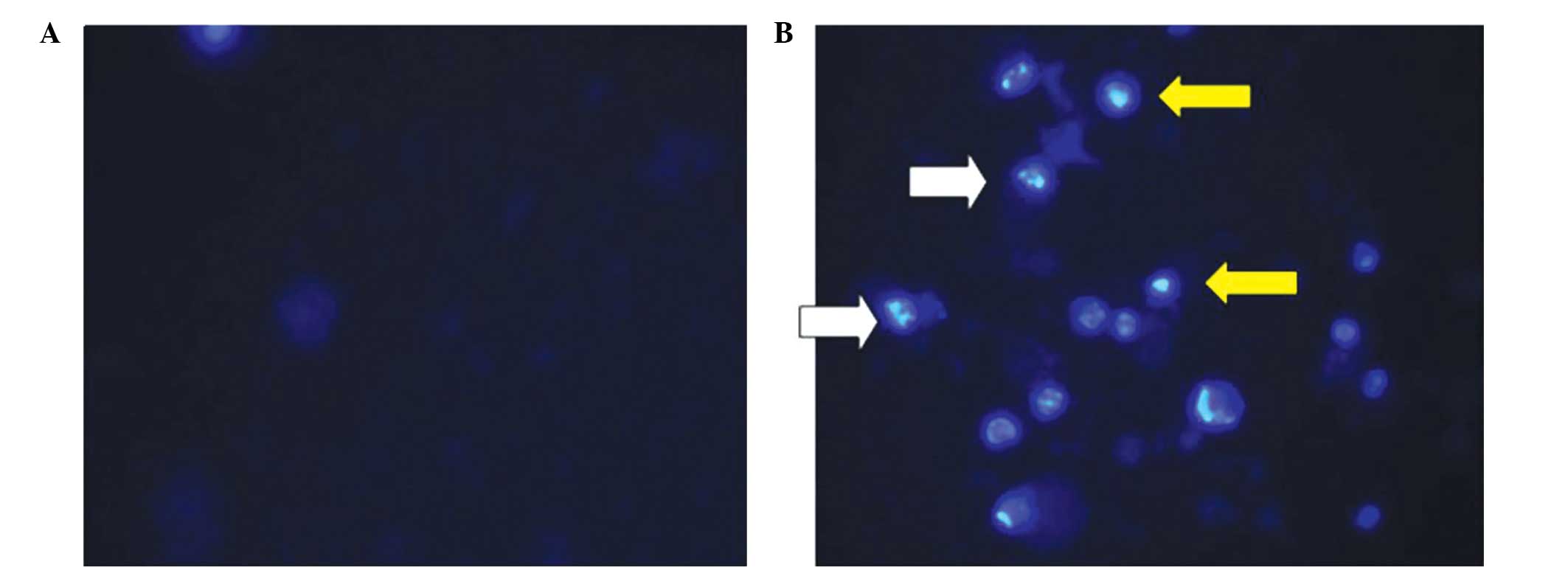
observations of the nuclear morphology revealed nuclear
condensation and DNA fragmentation in the APAP-treated Hep3B cells
(Fig. 5). These results overall
suggest that APAP can induce cell cytotoxicity via an increase in
the H2O2 level.
APAP activates the caspase-9/-3 cascade
in Hep3B cells
Caspase activation can induce cell apoptosis
(60,61). In our study, APAP treatment also
induced apoptosis of Hep3B cells, as indicated by results presented
in Figs. 4 and 5. Therefore, caspase activities were next
measured in Hep3B cells, focusing on the two major caspase
cascades, the caspase-9/-3 and the caspase-8/-3, and using a
substrate cleavage assay as previously described (60,61).
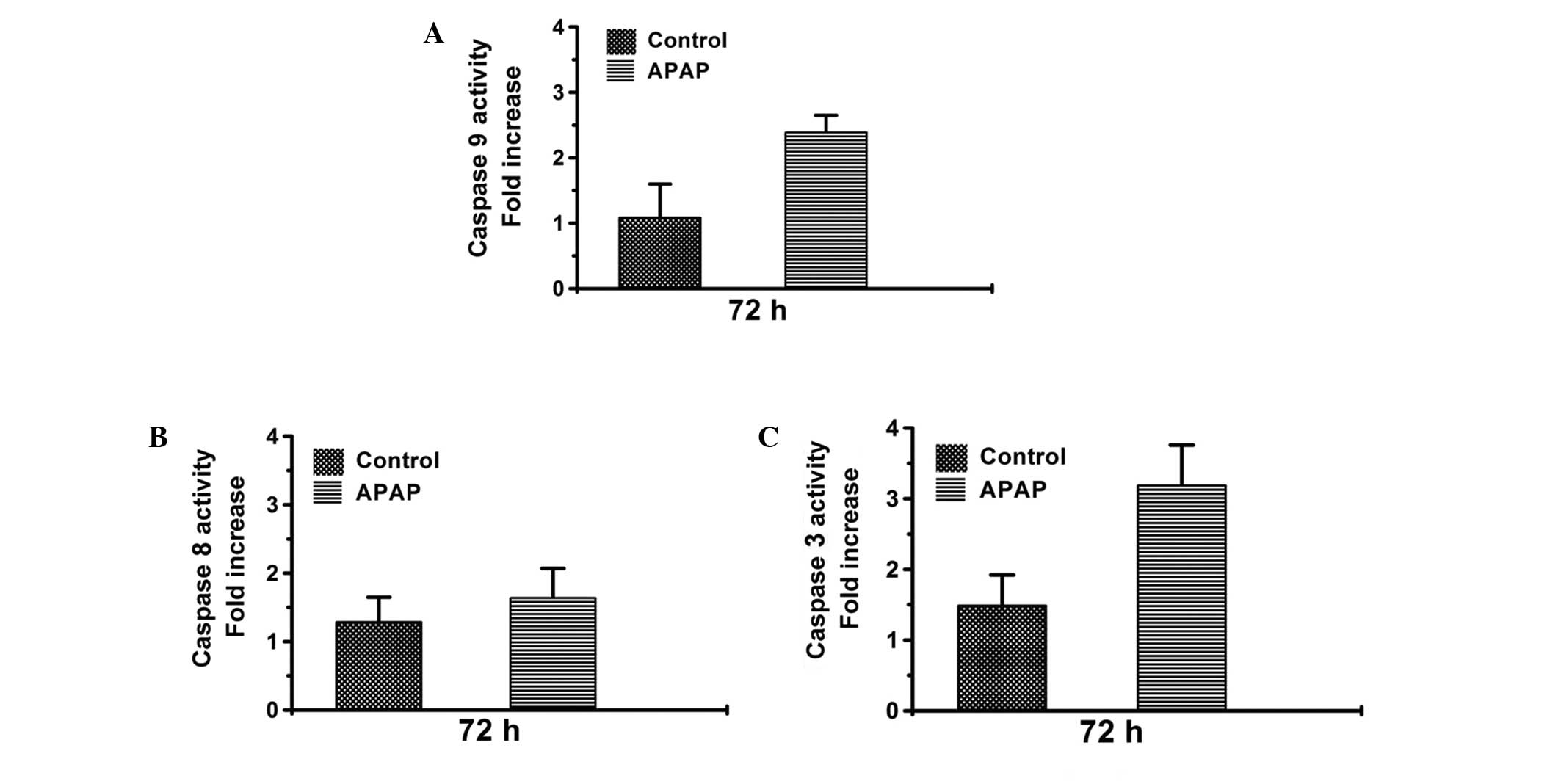
The caspase-9 and -3 activities were found induced by treatment
with 1/10 of the high dose of APAP (Fig. 6A and C) However, the caspase-8
activity did not notably change upon APAP treatment (Fig. 6B). This result suggests that APAP
can activate the caspase-9/-3 cascade to induce cell cytotoxicity
in Hep3B cells.
Discussion
Both tubular epithelial cell damage and fibroblast
proliferation can induce renal dysfunction (13,22,41,43,47,48,63,64).
Numerous studies have demonstrated that an APAP overdose can reduce
tubular epithelial cell survival, resulting in nephrotoxicity
(13,22,43–46).
Most of the studies to date have focused on high-dose APAP-induced
acute intoxication of kidney tubular cells. These studies have
highlighted the need to further investigate the effects of APAP and
take these effects into consideration in order to prevent
APAP-induced acute damage. However, it is still unclear whether low
doses of APAP may cause chronic kidney damage. Our present study
demonstrated that high-dose APAP treatment not only reduces
survival of tubular epithelial cells, but it can also induce
proliferation of fibroblasts, even at low doses. This implies that
APAP-induced renal damage may occur through epithelial cell damage
or fibroblast proliferation. In general, acute damage is easier to
detect and diagnose compared to chronic damage; therefore, APAP
overdose-induced acute intoxication is commonly observed, whereas
low-dose APAP-induced damage is more likely to be ignored in the
clinic. Here, we demonstrated that low-dose APAP treatment can
promote fibroblast proliferation. Thus, we consider the therapeutic
dose of APAP to be a safe analgesic and antipyretic agent for
patients who do not show fibrosis, but potentially harmful to
patients with kidney fibrosis.
The TGF-β signaling pathway was shown to be involved
in renal damage (67–69). TGF-β-induced renal damage has been
associated with: i) tubular cell death (68,70,71);
ii) epithelial mesenchymal transition (72,73);
and iii) fibroblast proliferation (74,75).
Up to now, no study has provided evidence that APAP can induce
kidney fibroblast proliferation via TGF-β-related signals. In this
study, NRK-49F cells (fibroblasts) treated with APAP showed a
similar induction in proliferation to the one observed in the group
treated with TGF-β. In addition, a previous study showed that TGF-β
is significantly elevated in APAP-treated liver tissue (71). Based on these observations, we
hypothesize that APAP induces kidney fibroblast proliferation via
the TGF-β signaling pathway. Whether APAP also exerts effects on
epithelial mesenchymal transition in kidney tubular cells warrants
future investigation.
O2− and
H2O2 are two commonly found ROS in the cells.
They are typically produced by the electron transport chain.
O2− can be removed from the cells through the
enzymatic activity of superoxide dismutase, and
H2O2 through the activity of catalase or
glutathione. It is well established that cell damage occurs when
O2− and H2O2 levels are
increased. Previous studies showed that an APAP overdose can
increase ROS levels and eventually, reduce cell viability (51,76).
However, these studies did not directly demonstrate which ROS
element is increased upon APAP treatment. Here, two types of ROS
(O2− and H2O2) were
quantified following APAP treatment. The H2O2
level increased, but no notable change in the
O2− level was observed in APAP-treated cells.
Our study suggests that the inhibition of cell survival by APAP may
occur through an increase in the H2O2 level.
This is possibly the reason why N-acetyl cysteine, a substrate for
glutathione synthesis, is applied on patients with APAP-induced
poisoning in emergency clinical cases (77,78).
APAP-induced cell death has been extensively studied
(13,22,44,47,48).
These studies demonstrated that APAP induces cell death either via
the apoptotic or the necrotic death pathways in different cells. In
our study, features of apoptosis were observed in APAP-treated
Hep3B cells, similar to previous studies (51,75).
Moreover, our study further demonstrated that the caspase-9/-3
cascade is activated upon APAP treatment, while the caspase-8/-3
cascade is not. Caspase-9/-3 signaling related to mitochondrial
damage and caspase-8/-3 signaling related to death receptor signals
have been previously reported (60,61).
Thus, our data suggest that APAP-induced cell cytotoxicity might be
associated with mitochondrial damage in Hep3B cells. Finally,
previous studies have shown cytotoxicity upon high-dose (>5 mM)
APAP treatment in vitro (15–20).
In this study, high-dose APAP treatment induced cytotoxicity in
both healthy kidney tubular cells and hepatoma cells. However, 1/10
of this dose was only cytotoxic to hepatoma cells. This suggests
that non-toxic (to healthy cells) doses of APAP may be applied in
the future as antitumor agents targeting cancer cells.
In summary, the present study shows that: i) APAP
treatment can induce cell proliferation of kidney fibroblasts even
at low doses, and thus we suggest that APAP treatment needs to be
carefully monitored in patients with fibrosis; ii) APAP treatment
can increase the H2O2 level and activate the
caspase-9/-3 cascade to cause cytotoxicity; and iii) the cytotoxic
effects of APAP depend on the cell type, with hepatoma cells being
more severely affected compared to healthy kidney tubular
cells.
Acknowledgements
This study was supported by the following grants:
NSC99-2320-B-039-030-MY3; NSC99-2632-B-039-001-MY3;
NSC101-2321-B-039-004; NHRI-EX102-10245BI; TCRD-I101-04-03;
TCRD-TPE-102-26; and TCRD-TPE-103-48.
References
|
1
|
Rumack BH: Acetaminophen misconceptions.
Hepatology. 40:10–15. 2004. View Article : Google Scholar
|
|
2
|
Cuzzolin L, Antonucci R and Fanos V:
Paracetamol (acetaminophen) efficacy and safety in the newborn.
Curr Drug Metab. 14:178–185. 2013.PubMed/NCBI
|
|
3
|
Klotz U: Paracetamol (acetaminophen) - a
popular and widely used nonopioid analgesic. Arzneimittelforschung.
62:355–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sullivan JE and Farrar HC: Section on
Clinical Pharmacology and Therapeutics, Committee on Drugs: Fever
and antipyretic use in children. Pediatrics. 127:580–587. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Young RJ: Dextropropoxyphene overdosage.
Pharmacological considerations and clinical management. Drugs.
26:70–79. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simkin S, Hawton K, Kapur N and Gunnell D:
What can be done to reduce mortality from paracetamol overdoses? A
patient interview study. QJM. 105:41–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hawton K, Bergen H, Simkin S, et al:
Impact of different pack sizes of paracetamol in the United Kingdom
and Ireland on intentional overdoses: a comparative study. BMC
Public Health. 11:4602011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hawton K, Townsend E, Deeks J, et al:
Effects of legislation restricting pack sizes of paracetamol and
salicylate on self poisoning in the United Kingdom: before and
after study. BMJ. 322:1203–1207. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Daly FF, Fountain JS, Murray L, Graudins A
and Buckley NA: Guidelines for the management of paracetamol
poisoning in Australia and New Zealand - explanation and
elaboration. A consensus statement from clinical toxicologists
consulting to the Australasian poisons information centres. Med J
Aust. 188:296–301. 2008.
|
|
10
|
Gopi KS, Reddy AG, Jyothi K and Kumar BA:
Acetaminophen-in duced hepato- and nephrotoxicity and amelioration
by silymarin and Terminalia chebula in rats. Toxicol Int.
17:64–66. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abdel-Zaher AO, Abdel-Hady RH, Mahmoud MM
and Farrag MM: The potential protective role of alpha-lipoic acid
against acetaminophen-induced hepatic and renal damage. Toxicology.
243:261–270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cermik H, Taslipinar MY, Aydin I, et al:
The relationship between N-acetylcysteine, hyperbaric oxygen, and
inflammation in a rat model of acetaminophen-induced
nephrotoxicity. Inflammation. 36:1145–1152. 2013. View Article : Google Scholar
|
|
13
|
Ucar F, Taslipinar MY, Alp BF, et al: The
effects of N-acetylcysteine and ozone therapy on oxidative stress
and inflammation in acetaminophen-induced nephrotoxicity model. Ren
Fail. 35:640–647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang YL, Zhang ZH, Liu XJ, et al:
Melatonin protects against apoptosis-inducing factor
(AIF)-dependent cell death during acetaminophen-induced acute liver
failure. PLoS One. 7:e519112012. View Article : Google Scholar
|
|
15
|
Amaral SS, Oliveira AG, Marques PE, et al:
Altered responsiveness to extracellular ATP enhances acetaminophen
hepatotoxicity. Cell Commun Signal. 11:102013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Badmann A, Langsch S, Keogh A, Brunner T,
Kaufmann T and Corazza N: TRAIL enhances paracetamol-induced liver
sinusoidal endothelial cell death in a Bim- and Bid-dependent
manner. Cell Death Dis. 3:e4472012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Badmann A, Keough A, Kaufmann T, Bouillet
P, Brunner T and Corazza N: Role of TRAIL and the pro-apoptotic
Bcl-2 homolog Bim in acetaminophen-induced liver damage. Cell Death
Dis. 2:e1712011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McGill MR, Yan HM, Ramachandran A, Murray
GJ, Rollins DE and Jaeschke H: HepaRG cells: a human model to study
mechanisms of acetaminophen hepatotoxicity. Hepatology. 53:974–982.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao X, Cong X, Zheng L, Xu L, Yin L and
Peng J: Dioscin, a natural steroid saponin, shows remarkable
protective effect against acetaminophen-induced liver damage in
vitro and in vivo. Toxicol Lett. 214:69–80. 2012. View Article : Google Scholar
|
|
20
|
Mobasher MA, Gonzalez-Rodriguez A,
Santamaria B, et al: Protein tyrosine phosphatase 1B modulates
GSK3beta/Nrf2 and IGFIR signaling pathways in acetaminophen-induced
hepatotoxicity. Cell Death Dis. 4:e6262013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ramachandran A, McGill MR, Xie Y, Ni HM,
Ding WX and Jaeschke H: Receptor interacting protein kinase 3 is a
critical early mediator of acetaminophen-induced hepatocyte
necrosis in mice. Hepatology. 58:2099–2108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ahmad ST, Arjumand W, Nafees S, et al:
Hesperidin alleviates acetaminophen induced toxicity in Wistar rats
by abrogation of oxidative stress, apoptosis and inflammation.
Toxicol Lett. 208:149–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inkielewicz-Stepniak I and Knap N: Effect
of exposure to fluoride and acetaminophen on oxidative/nitrosative
status of liver and kidney in male and female rats. Pharmacol Rep.
64:902–911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Slitt AM, Dominick PK, Roberts JC and
Cohen SD: Effect of ribose cysteine pretreatment on hepatic and
renal acetaminophen metabolite formation and glutathione depletion.
Basic Clin Pharmacol Toxicol. 96:487–494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yousef MI, Omar SA, El-Guendi MI and
Abdelmegid LA: Potential protective effects of quercetin and
curcumin on paracetamol-induced histological changes, oxidative
stress, impaired liver and kidney functions and haematotoxicity in
rat. Food Chem Toxicol. 48:3246–3261. 2010. View Article : Google Scholar
|
|
26
|
Zhang Y, Jia Y, Yang M, Yang P, Tian Y,
Xiao A and Wen A: The impaired disposition of probe drugs is due to
both liver and kidney dysfunctions in CCl(4)-model rats. Environ
Toxicol Pharmacol. 33:453–458. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao YL, Zhou GD, Yang HB, Wang JB, Shan
LM, Li RS and Xiao XH: Rhein protects against acetaminophen-induced
hepatic and renal toxicity. Food Chem Toxicol. 49:1705–1710. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roomi MW, Kalinovsky T, Ivanov V, Rath M
and Niedzwiecki A: A nutrient mixture prevents acetaminophen
hepatic and renal toxicity in ICR mice. Hum Exp Toxicol.
27:223–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Posadas I, Santos P and Cena V:
Acetaminophen induces human neuroblastoma cell death through NFKB
activation. PLoS One. 7:e501602012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Posadas I, Vellecco V, Santos P,
Prieto-Lloret J and Cena V: Acetaminophen potentiates
staurosporine-induced death in a human neuroblastoma cell line. Br
J Pharmacol. 150:577–585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jaeschke H: Comments on ‘glycogen synthase
kinase-3 mediates acetaminophen-induced apoptosis in human hepatoma
cells’. J Pharmacol Exp Ther. 314:1401–1404. 2005.
|
|
32
|
Macanas-Pirard P, Yaacob NS, Lee PC,
Holder JC, Hinton RH and Kass GE: Glycogen synthase kinase-3
mediates acetaminophen-induced apoptosis in human hepatoma cells. J
Pharmacol Exp Ther. 313:780–789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bilir A, Guneri AD and Altinoz MA:
Acetaminophen and DMSO modulate growth and gemcitabine cytotoxicity
in FM3A breast cancer cells in vitro. Neoplasma. 51:460–464.
2004.PubMed/NCBI
|
|
34
|
Wu YJ, Neuwelt AJ, Muldoon LL and Neuwelt
EA: Acetaminophen enhances cisplatin- and paclitaxel-mediated
cytotoxicity to SKOV3 human ovarian carcinoma. Anticancer Res.
33:2391–2400. 2013.PubMed/NCBI
|
|
35
|
Reszka KJ, Britigan LH, Rasmussen GT,
Wagner BA, Burns CP and Britigan BE: Acetaminophen stimulates the
peroxidative metabolism of anthracyclines. Arch Biochem Biophys.
427:16–29. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manov I, Bashenko Y, Eliaz-Wolkowicz A,
Mizrahi M, Liran O and Iancu TC: High-dose acetaminophen inhibits
the lethal effect of doxorubicin in HepG2 cells: the role of
P-glycoprotein and mitogen-activated protein kinase p44/42 pathway.
J Pharmacol Exp Ther. 322:1013–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kidokoro K, Satoh M, Nagasu H, et al:
Tacrolimus induces glomerular injury via endothelial dysfunction
caused by reactive oxygen species and inflammatory change. Kidney
Blood Press Res. 35:549–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stacchiotti A, Li Volti G, Lavazza A, et
al: Different role of Schisandrin B on mercury-induced renal damage
in vivo and in vitro. Toxicology. 286:48–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Suddek GM: Sunitinib improves
chemotherapeutic efficacy and ameliorates cisplatin-induced
nephrotoxicity in experimental animals. Cancer Chemother Pharmacol.
67:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Neria F, Castilla MA, Sanchez RF, et al:
Inhibition of JAK2 protects renal endothelial and epithelial cells
from oxidative stress and cyclosporin A toxicity. Kidney Int.
75:227–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jang HS, Kim JI, Jung KJ, Kim J, Han KH
and Park KM: Bone marrow-derived cells play a major role in kidney
fibrosis via proliferation and differentiation in the infiltrated
site. Biochim Biophys Acta. 1832.817–825. 2013.PubMed/NCBI
|
|
42
|
Seikrit C, Henkel C, van Roeyen CR, et al:
Biological responses to PDGF-AA versus PDGF-CC in renal
fibroblasts. Nephrol Dial Transplant. 28:889–900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim SH, Yu MA, Ryu ES, Jang YH and Kang
DH: Indoxyl sulfate-induced epithelial-to-mesenchymal transition
and apoptosis of renal tubular cells as novel mechanisms of
progression of renal disease. Lab Invest. 92:488–498. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Abdul Hamid Z, Budin SB, Wen Jie N, Hamid
A, Husain K and Mohamed J: Nephroprotective effects of Zingiber
zerumbet Smith ethyl acetate extract against
paracetamol-induced nephrotoxicity and oxidative stress in rats. J
Zhejiang Univ Sci B. 13:176–185. 2012.
|
|
45
|
Chen N, Aleksa K, Woodland C, Rieder M and
Koren G: The effect of N-acetylcysteine on ifosfamide-induced
nephrotoxicity: in vitro studies in renal tubular cells. Transl
Res. 150:51–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Isik B, Bayrak R, Akcay A and Sogut S:
Erdosteine against acetaminophen induced renal toxicity. Mol Cell
Biochem. 287:185–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lorz C, Justo P, Sanz A, Subira D, Egido J
and Ortiz A: Paracetamol-induced renal tubular injury: a role for
ER stress. J Am Soc Nephrol. 15:380–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lorz C, Justo P, Sanz AB, Egido J and
Ortiz A: Role of Bcl-xL in paracetamol-induced tubular epithelial
cell death. Kidney Int. 67:592–601. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Boulares AH, Zoltoski AJ, Stoica BA,
Cuvillier O and Smulson ME: Acetaminophen induces a
caspase-dependent and Bcl-XL sensitive apoptosis in human hepatoma
cells and lymphocytes. Pharmacol Toxicol. 90:38–50. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Manov I, Hirsh M and Iancu TC:
N-acetylcysteine does not protect HepG2 cells against
acetaminophen-induced apoptosis. Basic Clin Pharmacol Toxicol.
94:213–225. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Manov I, Hirsh M and Iancu TC:
Acetaminophen hepatotoxicity and mechanisms of its protection by
N-acetylcysteine: a study of Hep3B cells. Exp Toxicol Pathol.
53:489–500. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wu CS, Yen CJ, Chou RH, et al:
Cancer-associated carbohydrate antigens as potential biomarkers for
hepatocellular carcinoma. PLoS One. 7:e394662012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yu YL, Su KJ, Chen CJ, et al: Synergistic
anti-tumor activity of isochaihulactone and paclitaxel on human
lung cancer cells. J Cell Physiol. 227:213–222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang L, Li J, Jiang Z, et al: Inhibition
of aquaporin-1 expression by RNAi protects against aristolochic
acid I-induced apoptosis in human proximal tubular epithelial
(HK-2) cells. Biochem Biophys Res Commun. 405:68–73. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu YL, Chou RH, Wu CH, et al: Nuclear EGFR
suppresses ribonuclease activity of polynucleotide phosphorylase
through DNAPK-mediated phosphorylation at serine 776. J Biol Chem.
287:31015–31026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen KH, Li PC, Lin WH, Chien CT and Low
BH: Depression by a green tea extract of alcohol-induced oxidative
stress and lipogenesis in rat liver. Biosci Biotechnol Biochem.
75:1668–1676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin BR, Yu CJ, Chen WC, et al: Green tea
extract supplement reduces D-galactosamine-induced acute liver
injury by inhibition of apoptotic and proinflammatory signaling. J
Biomed Sci. 16:352009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yu YL, Yu SL, Su KJ, et al: Extended
O6-methylguanine methyltransferase promoter hypermethylation
following n-butylidenephthalide combined with
1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) on inhibition of human
hepatocellular carcinoma cell growth. J Agric Food Chem.
58:1630–1638. 2010. View Article : Google Scholar
|
|
59
|
Yu YL, Wei CW, Chen YL, Chen MH and Yiang
GT: Immunotherapy of breast cancer by single delivery with
rAAV2-mediated interleukin-15 expression. Int J Oncol. 36:365–370.
2010.PubMed/NCBI
|
|
60
|
Yiang GT, Chen YH, Chou PL, Chang WJ, Wei
CW and Yu YL: The NS3 protease and helicase domains of Japanese
encephalitis virus trigger cell death via caspase-dependent and
-independent pathways. Mol Med Rep. 7:826–830. 2013.PubMed/NCBI
|
|
61
|
Yiang GT, Yu YL, Hu SC, Chen MH, Wang JJ
and Wei CW: PKC and MEK pathways inhibit caspase-9/-3-mediated
cytotoxicity in differentiated cells. FEBS Lett. 582:881–885. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wei CW, Lin CC, Yu YL, et al:
n-Butylidenephthalide induced apoptosis in the A549 human lung
adenocarcinoma cell line by coupled down-regulation of AP-2alpha
and telomerase activity. Acta Pharmacol Sin. 30:1297–1306. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hassane S, Leonhard WN, van der Wal A, et
al: Elevated TGFbeta-Smad signalling in experimental Pkd1 models
and human patients with polycystic kidney disease. J Pathol.
222:21–31. 2010.PubMed/NCBI
|
|
64
|
Edward M, Quinn JA, Mukherjee S, et al:
Gadodiamide contrast agent ‘activates’ fibroblasts: a possible
cause of nephrogenic systemic fibrosis. J Pathol. 214:584–593.
2008.
|
|
65
|
Kumari A and Kakkar P: Lupeol protects
against acetaminophen-induced oxidative stress and cell death in
rat primary hepatocytes. Food Chem Toxicol. 50:1781–1789. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Anoush M, Eghbal MA, Fathiazad F, Hamzeiy
H and Kouzehkonani NS: The protective effects of garlic extract
against acetaminophen-induced oxidative stress and glutathione
depletion. Pak J Biol Sci. 12:765–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yao Y, Yang J, Wang D, et al: The aqueous
extract of Lycopus lucidus Turcz ameliorates
streptozotocin-induced diabetic renal damage via inhibiting
TGF-beta1 signaling pathway. Phytomedicine. 20:1160–1167. 2013.
|
|
68
|
Hsieh TJ, Hsieh PC, Tsai YH, et al:
Melamine induces human renal proximal tubular cell injury via
transforming growth factor-beta and oxidative stress. Toxicol Sci.
130:17–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hills CE, Siamantouras E, Smith SW,
Cockwell P, Liu KK and Squires PE: TGFβ modulates cell-to-cell
communication in early epithelial-to-mesenchymal transition.
Diabetologia. 55:812–824. 2012.
|
|
70
|
Xu Y, Yang S, Huang J, Ruan S, Zheng Z and
Lin J: Tgf-β1 induces autophagy and promotes apoptosis in renal
tubular epithelial cells. Int J Mol Med. 29:781–790. 2012.
|
|
71
|
Yoshikawa M, Hishikawa K, Idei M and
Fujita T: Trichostatin a prevents TGF-beta1-induced apoptosis by
inhibiting ERK activation in human renal tubular epithelial cells.
Eur J Pharmacol. 642:28–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li R, Wang Y, Liu Y, et al: Curcumin
inhibits transforming growth factor-β1-induced EMT via PPARγ
pathway, not Smad pathway in renal tubular epithelial cells. PLoS
One. 8:e588482013.
|
|
73
|
Han WQ, Zhu Q, Hu J, Li PL, Zhang F and Li
N: Hypoxia-inducible factor prolyl-hydroxylase-2 mediates
transforming growth factor beta 1-induced epithelial-mesenchymal
transition in renal tubular cells. Biochim Biophys Acta.
1833.1454–1462. 2013.
|
|
74
|
Yan HD, Li XZ, Xie JM and Li M: Effects of
advanced glycation end products on renal fibrosis and oxidative
stress in cultured NRK-49F cells. Chin Med J (Engl). 120:787–793.
2007.
|
|
75
|
Guo W, Xu H, Huang WY, et al: Prohibitin
suppresses renal interstitial fibroblasts proliferation and
phenotypic change induced by transforming growth factor-beta1.
Zhonghua Yi Xue Za Zhi. 87:1660–1665. 2007.(In Chinese).
|
|
76
|
Levanon D, Manov I and Iancu TC:
Qualitative and quantitative analysis of the effects of
acetaminophen and N-acetylcysteine on the surface morphology of
Hep3B hepatoma cells in vitro. Ultrastruct Pathol. 28:3–14.
2004.PubMed/NCBI
|
|
77
|
Williamson K, Wahl MS and Mycyk MB: Direct
comparison of 20-hour IV, 36-hour oral, and 72-hour oral
acetylcysteine for treatment of acute acetaminophen poisoning. Am J
Ther. 20:37–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Saritas A, Kandis H, Baltaci D, et al:
N-Acetyl cysteine and erdosteine treatment in acetaminophen-induced
liver damage. Toxicol Ind Health. Oct 15–2012.(Epub ahead of
print).
|