Introduction
Angiogenesis is a complex multi-step process that
occurs in response to ischemic stimuli. These steps include
degradation of the basement membrane, proliferation, apoptosis and
invasion of endothelial cells, tube formation, and anastomosis
(1,2). Various proteases including the family
of matrix metalloproteinases (MMPs), and cysteine and serine
proteases contribute to the complex processes of new vessel
formation and remodeling. Urbich et al (3) demonstrated that cysteine protease
cathepsin L-deficient mice presented impaired wound recovery,
indicating an important role for cathepsin L in neovascularization.
Furthermore, the study validated the ability of cathepsin L to
induce angiogenesis, as transferred cathepsin L-deficient
progenitor cells did not migrate to ischemic areas or augment
vasculogenesis. Conversely, forced expression of cathepsin L in
mature endothelial cells markedly enhanced cell invasion.
Furthermore, Shi et al (4)
demonstrated that cathepsin S was required for microvessel
formation. Cysteine proteases have previously been demonstrated to
be important in apoptosis and cell survival, separate from their
role in proteolysis of extracellular matrix in vascular remodeling
(5–7); however, the underlying mechanism of
this process remains to be fully elucidated.
Angiogenesis is associated with atherosclerosis, and
numerous risk factors of atherosclerosis, including diabetes and
insulin resistance, are accompanied by high levels of free fatty
acids (FFA) (8,9). However, the specific role of FFAs and
their association with angiogenesis remains to be elucidated. FFAs
have been demonstrated to exert effects on endothelial cells via
enhancing reactive oxygen species levels or impairing nitric oxide
production (10); therefore,
increased levels of FFA will inhibit angiogenesis. Palmitate, which
is the most frequently occurring form of saturated FFA present in
human serum, contributes to lipotoxicity (11). In addition to the aforementioned
characteristics, palmitate has been detected to induce apoptosis in
a variety of tissues (10,12,13)
and decrease cardiolipid synthesis, resulting in the release of
cytochrome c (14).
Cathepsin L and S have therefore been confirmed to
be important in endothelial cell angiogenesis; however, it remains
to be elucidated as to whether FFA levels may influence
cathepsin-mediated angiogenesis. The present study examined the
proliferation, apoptosis and invasion of human umbilical vein
endothelial cells (HUVECs) following exposure to palmitate in the
presence or absence of selective cathepsin inhibitors, and observed
that palmitate impaired cathepsin protein expression levels and
activity.
Materials and methods
Cell culture and incubation with fatty
acids
HUVECs were purchased from the American Type Culture
Collection (Manassas, VA, USA; PCS-100-010) and cultured in M199
medium (HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 20% fetal bovine serum (FBS; HyClone; GE
Healthcare Life Sciences) at 37°C in an atmosphere containing under
5% CO2. Cells were passaged every 2–3 days once they
reached maximum confluence. Cells were incubated in M199/10% FBS
medium supplemented with 0.05, 0.1, 0.2, 0.4 or 0.6 mM palmitate
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) preconjugated with
FFA-free bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) at a
1:1 molar ratio. Control cells were grown with the same medium
containing FFA-free BSA. If not stated otherwise, cells were
incubated for 1 h with 10 µΜ cathepsin L inhibitor (z-FF-FMK; cat.
no. 219421; Calbiochem; EMD Millipore, Billerica, MA, USA) and
cathepsin S inhibitor (z-FL-COCHO.H2O; cat. no. 219393;
Calbiochem; EMD Millipore) at 37°C, which was followed by
incubation with palmitate or FFA-free BSA for 24 h at 37°C.
Immunofluorescence staining
HUVECs were fixed in 4% paraformaldehyde for 20 min
and incubated at 37°C in blocking buffer (PBS containing 5% BSA).
Cells were incubated in the presence of mouse anti-Cluster of
Differentiation (CD) 31 antibody (1:200; cat. no. SC-81158; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) for 2 h at 37°C and
washed three times in PBS. Cells were subsequently incubated with
rhodamine-conjugated goat anti-mouse IgG (H+L) secondary antibody
(1:1,000; cat. no. 31660; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) for 1 h at 37°C. Nuclei were stained with DAPI (1:10,000;
Invitrogen; Thermo Fisher Scientific, Inc.) and were examined with
an Olympus IX70 inverted fluorescence microscope.
Cell proliferation assay
The effect of elevated palmitate concentration on
HUVEC proliferation was analyzed utilizing Cell Counting kit
(CCK)-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
according to the manufacturer's protocol. The amount of the
formazan dye generated in cells was directly proportional to the
number of living cells. Absorbance of the samples was measured at a
wavelength of 450 nm.
Cell apoptosis assay
HUVECs were grown in M199 medium and pretreated for
1 h at 37°C with indicated protease inhibitors prior to the
addition of 0, 0.05, 0.1, 0.2, 0.4 or 0.6 mM palmitate. Following
palmitate treatment for 24 h at 37°C, cells were harvested. A total
of 1×106 cells were suspended in 200 µl Annexin V
binding buffer (BD Biosciences, Franklin Lakes, NJ, USA) and
incubated with 2.5 µl annexin V-fluorescein isothiocyanate (FITC)
and 2.5 µl propidium iodide (PI; BD Biosciences) for 15 min at room
temperature in the dark. Unstained cells and those stained with
Annexin V-FITC or PI alone were used as controls to set up
compensation and quadrants. Cell apoptosis was investigated by flow
cytometry within 30 min after fluorescent labeling by using
FlowJo® software (version 8.8.7; Tree Star, Inc.,
Ashland, OR, USA). Results were determined as the percentage of
cells that were early (Annexin V+PI−) or late
(Annexin V+PI+) apoptotic.
In vitro invasion
HUVEC invasion across a polycarbonate membrane
containing 8-µm pores was performed with Transwell 24-well plates
according to the manufacturer's protocol. Briefly, the membrane was
precoated with type I collagen (1 mg/ml; Sigma-Aldrich; Merck
KGaA). Detached cells (1×105 cells per 100 µl in
FBS-free M199 containing 0, 0.05, 0.2, 0.4 and 0.6 mM palmitate)
were placed in the upper chambers. The lower chamber contained 600
µl of M199 supplemented with 20% FBS and cells were incubated for
16 h at 37°C. Following incubation, cells that remained on the
upper surface of the membrane were removed by wiping with a cotton
swab, while cells that crossed onto the lower side of the membrane
were fixed with 4% paraformaldehyde for 30 min at room temperature
and subsequently stained with crystal violet for 20 min at room
temperature. The number of cells was counted manually in six random
microscope fields by two independent investigators. The experiments
were repeated in the presence of 10 µΜ cathepsin inhibitors for 1 h
prior to the addition of palmitate, as described above for cell
culture.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted using TRIzol®
reagent (Sigma-Aldrich; Merck KGaA). cDNA was synthesized from 2 µg
total RNA using ReverTra Ace® qPCR Reverse Transcriptase
kit (cat. no. FSQ-101; Toyobo Co., Ltd., Osaka, Japan) according to
the manufacturer's protocol. qPCR was performed using the
SYBR-Green Master mix (cat. no. QPK-212; Toyobo Co., Ltd.) in an
ABI 7500HT Real-Time PCR machine (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions for PCR were 95°C
for 60 sec, followed by 40 cycles at 95°C for 15 sec, 61°C for 15
sec and 72°C for 45 sec. The relative expression level was
determined by the 2−∆∆Cq method and normalized against
GAPDH (15). The primer sequences
used were as follows: Forward, 5′-CGAATCATTGAAGATCCGAGTG-3′ and
reverse, 5′-ATGGCTTAGAGCCCAATTATGT-3′ for cathepsin L; forward,
5-TAT TGC CTG ATT CTG TGG ACT G-3 and reverse, 5-TGA TGT ACTG GAA
AGC CGT TGT-3 for cathepsin S; forward,
5′-GCAGATCGTAGCTGGGGTGAACT-3′ and reverse,
5′-AAGCAAGAAGGAAGGAGGGAGGG-3′ for cystatin C; and forward,
5′-TGCACCACCAACTGCTTAGC-3′ and reverse, 5′-GGCATGGACTGTGGTCATGAG-3′
for GAPDH.
Western blot analysis
Cells were lysed in ice-cold
radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck KGaA)
supplemented with 1 mM phenomethylsulfonyl fluoride (Thermo Fisher
Scientific, Inc.). The proteins (20 µg per lane) were separated on
a 12% SDS-PAGE gel (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and electrotransferred onto a polyvinylidene difluoride membrane.
Following transfer, blocking was performed with 5% BSA
(Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. Membranes
were probed with specific primary antibodies at 4°C overnight and
horseradish peroxidase (HRP)-conjugated anti-rabbit/mouse
immunoglobulin secondary antibodies (1:5,000; cat. nos. SC-2004 and
SC-2005; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. The primary antibodies used were mouse anti-cathepsin
L (1:1,000; cat. no. ab6314; Abcam, Cambridge, UK), rabbit
anti-cathepsin S (1:5,000; cat. no. ab50400; Abcam), rabbit
anti-cystatin C (1:500; cat. no. ab33487; Abcam) and mouse
anti-tubulin (1:1,000; cat. no. AT819; Beyotime Institute of
Biotechnology, Haimen, China). Enhanced chemiluminescence was
performed using the Pro-light HRP Chemiluminescence kit according
to the instructions of the manufacturer (Tiangen Biotech Co., Ltd.,
Beijing, China). Membranes were scanned and visualized by an
LAS3000 system (LAS3000 mini ImageReader, Fuji Photo Film Co.,
Ltd., Japan).
Cathepsins L and S activity assay
Cells were lysed and treated with reaction buffer
that was included in the kits used for cathepsin L (cat. no.
ab65306; Abcam) or cathepsin S (cat. no. ab65307; Abcam), and 10 mM
fluorogenic Ac-FR-amino-4-trifluoromethyl coumarin (AFC) substrate,
which is the preferred cathepsin L substrate (cat. no. ab65306;
Abcam) or 10 mM Ac-VVR-AFC, which is the preferred cathepsin S
substrate (cat. no. ab65307; Abcam) according to the manufacturer's
protocol. Fluorescence was measured using SpectraMax M5
fluorometer, at an excitation wavelength of 400 nm and an emission
wavelength of 505 nm.
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. Multiple group
comparisons were performed by one-way analysis of variance followed
by the least significant difference post hoc test. All tests
analyzed were two-sided. P<0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
conducted using SPSS software version 11.5 (SPSS, Inc., Chicago,
IL, USA).
Results
Effect of palmitate and cysteine
protease on HUVEC proliferation
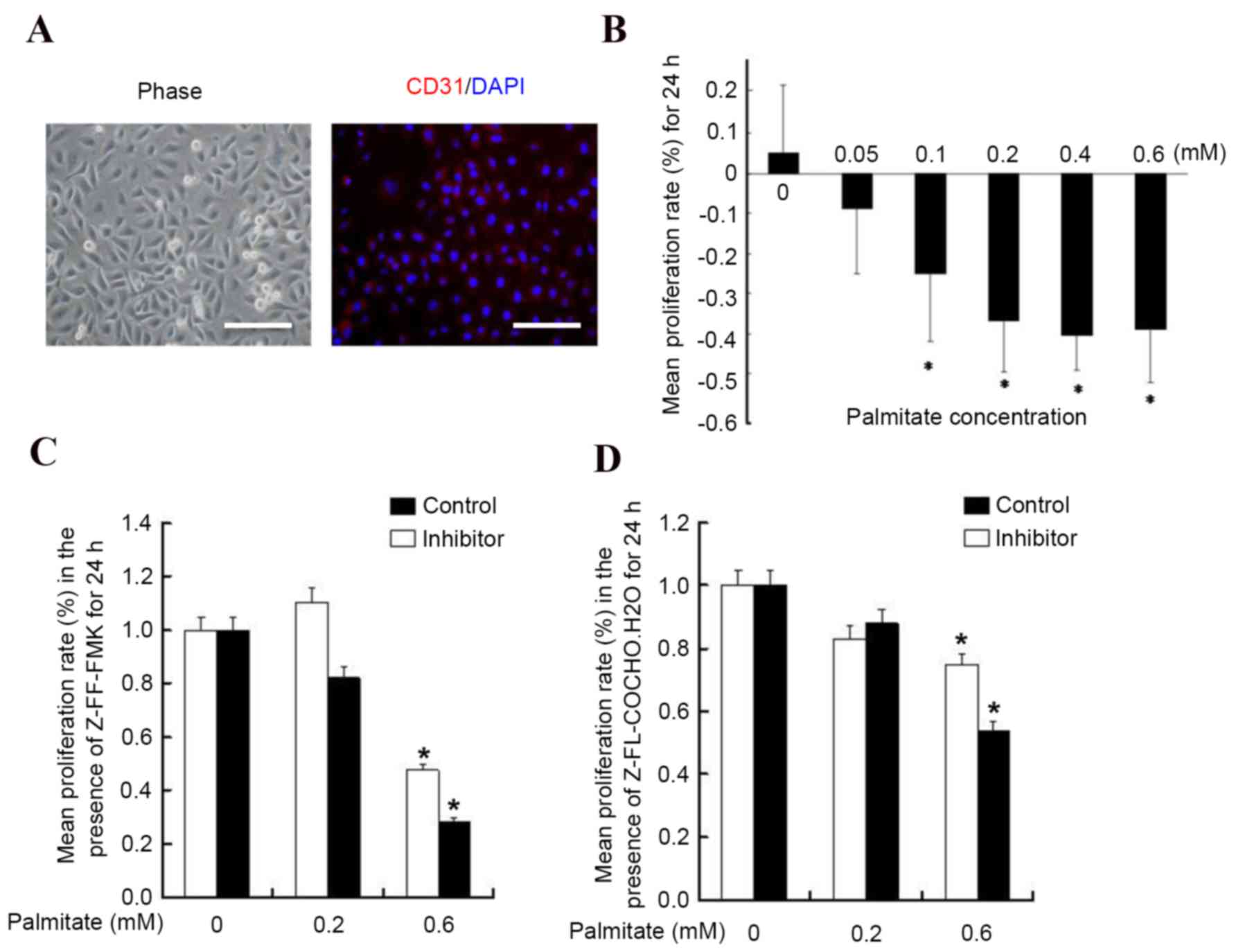
HUVECs express the endothelial marker protein CD31
as previously described (16), and
as presented in Fig. 1A. To
examine the effect of palmitate on HUVEC proliferation, HUVECs were
incubated with different concentrations of palmitate and it was
observed that palmitate decreased HUVEC proliferation in a
dose-dependent manner (Fig. 1B).
High levels of palmitate (0.6 mM) inhibited the proliferation rate
significantly by 44±7% (P<0.05 vs. 0 mM), whereas the lowest
concentration (0.05 mM) had no significant effect (P=0.09 vs. 0
mM). These results are consistent with a previous report (17). To determine whether cathepsin is
associated with HUVEC proliferation in the presence of a high
concentration of FFA, cysteine protease inhibitors were added to
the concentrations of palmitate. It was observed that treatment
with the cathepsin L inhibitor Z-FF-FMK (Fig. 1C) and the cathepsin S inhibitor
Z-FL-COCHO (Fig. 1D) did not
result in significant alterations of the growth rate (P>0.05),
suggesting palmitate reduced HUVEC proliferation via a
cathepsin-independent pathway.
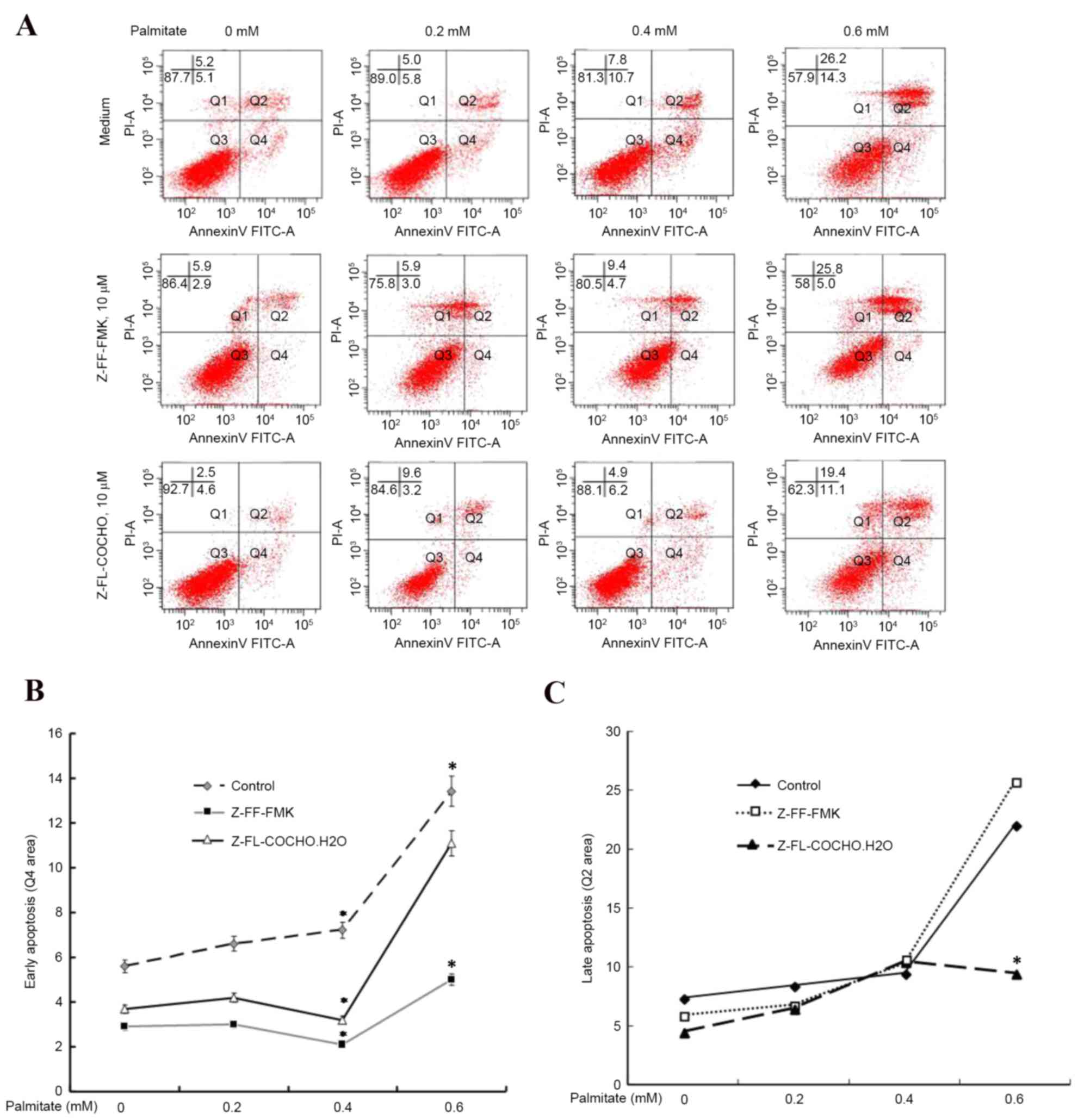
Cysteine protease inhibitors attenuate
palmitate-induced HUVEC apoptosis
Palmitate may trigger apoptotic pathways in HUVECs
(17); therefore, the present
study examined whether cathepsin regulated palmitate-induced HUVEC
apoptosis (Fig. 2A). As presented
in Fig. 2B, cathepsin L and S
inhibitors exhibited the ability to reduce the early apoptosis of
HUVECs induced by palmitate by almost 50%, suggesting a key
cathepsin-dependent pathway in palmitate-induced apoptosis.
Furthermore, cathepsin L inhibitor Z-FF-FMK exhibited the ability
to reduce early apoptosis more effectively compared with the
cathepsin S inhibitor Z-FL-COCHO. However, late apoptosis appeared
to be unaffected by cathepsin inhibitors (Fig. 2C). These results suggested that
cathepsin L and S are associated with cell apoptosis as the
cathepsin inhibitors protected cells from high FFA level-induced
apoptosis.
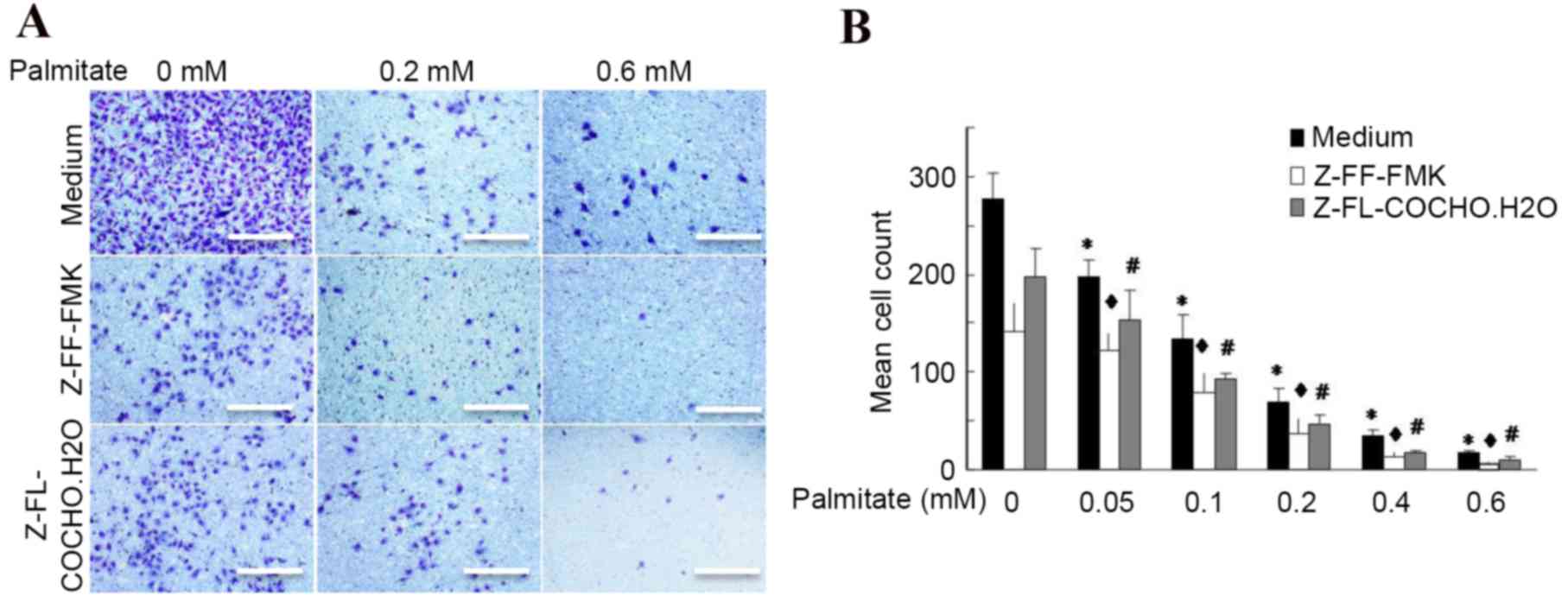
Palmitate inhibits cathepsin-mediated
cell invasion
Previous reports have demonstrated that cathepsin L
is required for endothelial progenitor cell invasion and
neovascularization (3,18); however, whether other cathepsins
may influence cell invasion and whether the effects of decreased
invasion result from increased FFA levels remains to be elucidated.
The present study firstly assessed the effect of palmitate on cell
invasion ability using a Transwell assay. It was observed that 0.4
and 0.6 mM palmitate reduced endothelial cell invasion by 88 and
94%, respectively, compared with untreated cells (Fig. 3), indicating that palmitate
inhibits cell invasion in a dose-dependent manner. Furthermore,
cathepsin S and L inhibitors significantly reduced the cell
migration by almost 2-fold, even in the presence of a high level of
palmitate (Fig. 3). These findings
indicated that cathepsins contribute to cell migration and invasion
and palmitate is a potential factor that may reduce
cathepsin-mediated cell invasion.
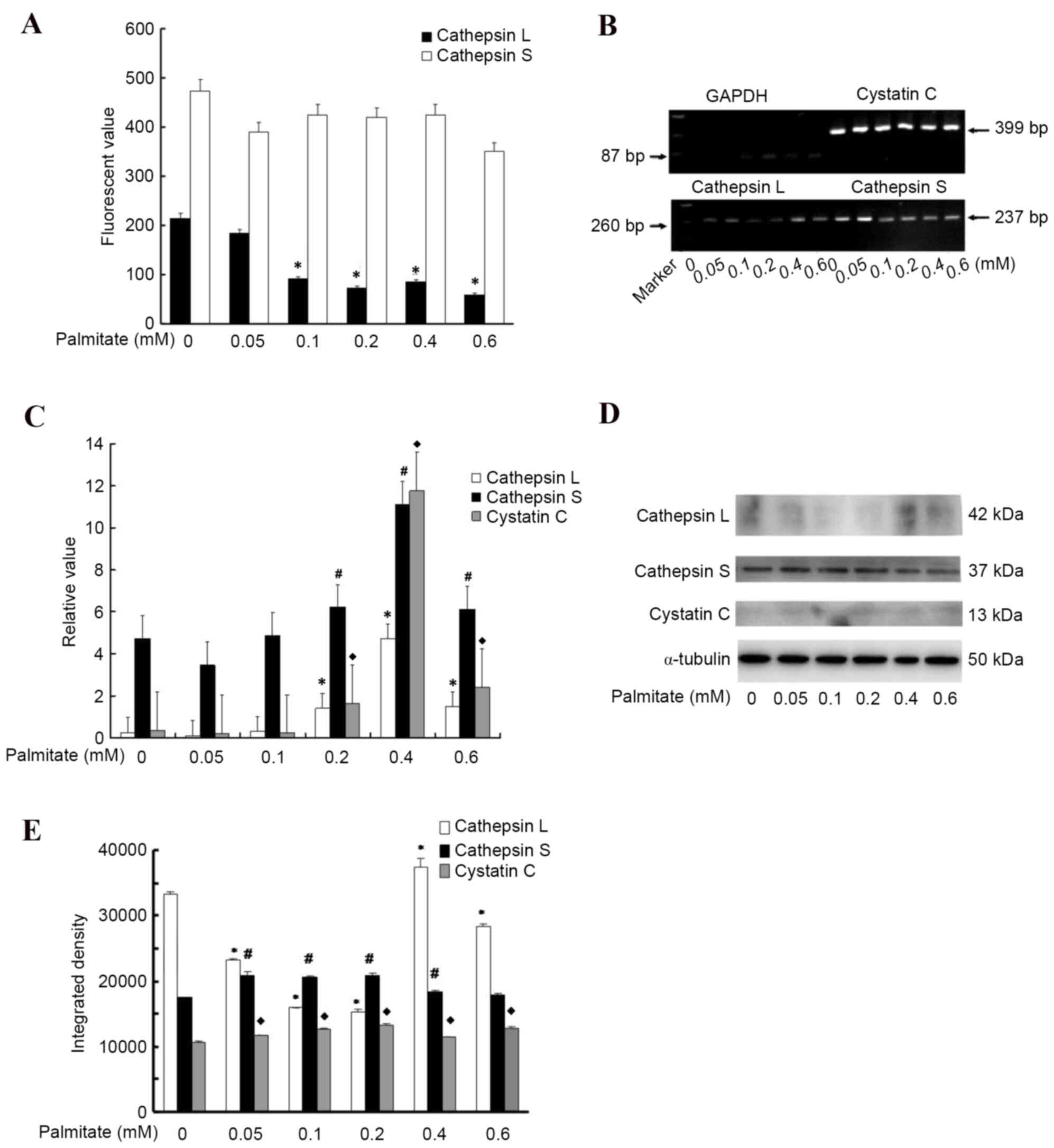
Palmitate suppresses the activity of
cathepsins L and S
To investigate the underlying mechanism of
cathepsins L and S reduction in response to high FFA levels, the
present study measured the effect of high palmitate levels on the
mRNA and protein expression levels, and activities of cathepsin L
and S in HUVECs. The protease activity experiment demonstrated that
cells incubated in palmitate for 24 h presented a markedly
attenuated cathepsin L activity (P<0.05; Fig. 4A) compared with those cells
cultured in medium without palmitate. The greatest palmitate level
decreased the activity of cathepsin L by almost 3.6 fold (Fig. 4A). In addition, the high palmitate
level attenuated cathepsin S activity; however, this alteration was
not significant (Fig. 4A). To
further analyze the effect of high palmitate levels on protease
expression, the present study detected the mRNA expression levels
of cathepsin L, S and their natural inhibitor cystatin C, and
observed that palmitate an increased expression levels in a
dose-dependent manner (Fig. 4B and
C), suggesting a regulatory interaction between palmitate and
cathepsins or their endogenous inhibitor. Consistent with the mRNA
expression, cathepsin L protein level was upregulated by high
palmitate levels at 0.4 mM but was downregulated by 0.6 mM
palmitate (Fig. 4D and E), thus,
high palmitate levels (>0.4 mM) may have a role in inhibiting
cathepsin L activity. However, the protein expression levels of
cathepsin S and cystatin C appeared to vary only slightly with
different palmitate concentrations, the differences between the
mRNA and protein results may be a result of posttranscriptional
modifications.
Discussion
The present study demonstrated that exposure of
cultured HUVECs to palmitate led to impairment of cell
proliferation and invasion, and increased cell apoptosis, and that
the function of palmitate is associated with cathepsin L and S.
Further experiments demonstrated that palmitate inhibited the
activity of cathepsin L and S in HUVECs.
The effects of FFA on cell proliferation, apoptosis
and invasion have previously been investigated; Listenberger et
al (10) observed that
saturated fatty acids increase nicotinamide adenine dinucleotide
phosphate oxidase-generated oxidant stress, nuclear factor-kB
activation and ceramide accumulation. The latter, which may be
synthesized de novo from saturated fatty acids and the amino
acid serine, has been implicated in apoptosis and oxidative stress
modulation (19–21). Consistent with these reports, high
concentrations of saturated fatty acids may trigger apoptosis and
inhibit cell cycle progression in cultured human endothelial cell
monolayers (22–24). In addition, trophoblast cells
treated with long-chain fatty acids revealed increased lipid
droplet deposition, severe mitochondrial damage and significantly
decreased invasion (25). The
present study demonstrated that the FFA palmitate exhibits similar
effects on HUVECs.
Cysteine proteases contribute to angiogenesis under
pathophysiological conditions. Cathepsins were originally
identified as members of the cysteine protease family localized in
lysosomes. However, previous data revealed unexpected roles for
cathepsins in pathological conditions including cell survival,
metabolic disorder and atherosclerosis-based cardiovascular disease
(26–31). However, the definite underlying
molecular mechanism remains to be fully elucidated. A variety of
cathepsins (B, D, L, S and K) have been demonstrated to cleave the
pro-apoptotic B-cell lymphoma-2 (Bcl-2) family member BH3
interacting domain death agonist (Bid) into its potent
pro-apoptotic tBid fragment in vitro (32,33).
Translocation of these pro-apoptotic fragments to the mitochondrial
outer membrane may induce the release of apoptogenic factors
including cytochrome c and subsequent activation of downstream
caspases. In addition to Bcl-2 family members, caspases may be
appropriate candidates as cathepsin substrates (34–39).
It was therefore hypothesized that cysteine proteases contribute to
the regulation of apoptotic signaling mechanisms. Consistent with
this, the present study observed that cathepsin inhibitors
protected cells from high FFA level-induced apoptosis. In addition,
it has previously been demonstrated that intracellular and
extracellular cathepsin S, L and K activities were reduced in
non-invasive compared with highly invasive cells (40), and downregulation of cathepsins L
and S led to defective endothelial cell invasion and suppression of
proliferation (41,42), indicating that cathepsins are
important for angiogenesis. Therefore, cathepsins S and L are
required for endothelial progenitor cell-induced neovascularization
in response to ischemic stress (3,43).
In conclusion, the results of the present study, in
conjunction with previous findings, support the anti-angiogenic
action of FFA. The results suggested that the cathepsin-dependent
effects should be considered when developing novel therapies during
human endothelial cell angiogenesis. These findings suggested a
novel approach to pharmacological and genetic rescue strategies in
FFA-mediated angiogenesis.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81300167 and 30873350).
References
|
1
|
Binet F and Sapieha P: ER stress and
angiogenesis. Cell Metab. 22:560–575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pourrajab F, Zarch A Vakili,
Hekmatimoghaddam S and Zare-Khormizi MR: MicroRNAs; easy and potent
targets in optimizing therapeutic methods in reparative
angiogenesis. J Cell Mol Med. 19:2702–2714. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Urbich C, Heeschen C, Aicher A, Sasaki K,
Bruhl T, Farhadi MR, Vajkoczy P, Hofmann WK, Peters C, Pennacchio
LA, et al: Cathepsin L is required for endothelial progenitor
cell-induced neovascularization. Nat Med. 11:206–213. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi GP, Sukhova GK, Kuzuya M, Ye Q, Du J,
Zhang Y, Pan JH, Lu ML, Cheng XW, Iguchi A, et al: Deficiency of
the cysteine protease cathepsin S impairs microvessel growth. Circ
Res. 92:493–500. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Conus S and Simon HU: Cathepsins: Key
modulators of cell death and inflammatory responses. Biochem
Pharmacol. 76:1374–1382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Droga-Mazovec G, Bojic L, Petelin A,
Ivanova S, Romih R, Repnik U, Salvesen GS, Stoka V, Turk V and Turk
B: Cysteine cathepsins trigger caspase-dependent cell death through
cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem.
283:19140–19150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng X, Chu F, Mirkin BL, Sudha T, Mousa
SA and Rebbaa A: Role of the proteolytic hierarchy between
cathepsin Lcathepsin D and caspase-3 in regulation of cellular
susceptibility to apoptosis and autophagy. Biochim Biophys Acta.
1783:2294–2300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shulman GI: Cellular mechanisms of insulin
resistance. J Clin Invest. 106:171–176. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boden G and Shulman GI: Free fatty acids
in obesity and type 2 diabetes: Defining their role in the
development of insulin resistance and beta-cell dysfunction. Eur J
Clin Invest. 32 Suppl 3:S14–S23. 2002. View Article : Google Scholar
|
|
10
|
Listenberger LL, Ory DS and Schaffer JE:
Palmitate-induced apoptosis can occur through a
ceramide-independent pathway. J Biol Chem. 276:14890–14895. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Listenberger LL, Han X, Lewis SE, Cases S,
RV Jr, Ory DS Farese and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:pp. 3077–3082. 2003; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paumen MB, Ishida Y, Muramatsu M, Yamamoto
M and Honjo T: Inhibition of carnitine palmitoyltransferase I
augments sphingolipid synthesis and palmitate-induced apoptosis. J
Biol Chem. 272:3324–3329. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cnop M, Hannaert JC, Hoorens A, Eizirik DL
and Pipeleers DG: Inverse relationship between cytotoxicity of free
fatty acids in pancreatic islet cells and cellular triglyceride
accumulation. Diabetes. 50:1771–1777. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ostrander DB, Sparagna GC, Amoscato AA,
McMillin JB and Dowhan W: Decreased cardiolipin synthesis
corresponds with cytochrome c release in palmitate-induced
cardiomyocyte apoptosis. J Biol Chem. 276:38061–38067.
2001.PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pinter E, Mahooti S, Wang Y, Imhof BA and
Madri JA: Hyperglycemia-induced vasculopathy in the murine
vitelline vasculature: Correlation with PECAM-1/CD31 tyrosine
phosphorylation state. Am J Pathol. 154:1367–1379. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ciapaite J, Van Bezu J, Van Eikenhorst G,
Bakker SJ, Teerlink T, Diamant M, Heine RJ, Krab K, Westerhoff HV
and Schalkwijk CG: Palmitate and oleate have distinct effects on
the inflammatory phenotype of human endothelial cells. Biochim
Biophys Acta. 1771:147–154. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Urbich C, Dernbach E, Rössig L, Zeiher AM
and Dimmeler S: High glucose reduces cathepsin L activity and
impairs invasion of circulating progenitor cells. J Mol Cell
Cardiol. 45:429–436. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shimabukuro M, Zhou YT, Levi M and Unger
RH: Fatty acid-induced beta cell apoptosis: A link between obesity
and diabetes. Proc Natl Acad Sci USA. 95:pp. 2498–2502. 1998;
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia
R, Herrero P, Saffitz JE and Schaffer JE: A novel mouse model of
lipotoxic cardiomyopathy. J Clin Invest. 107:813–822. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gudz TI, Tserng KY and Hoppel CL: Direct
inhibition of mitochondrial respiratory chain complex III by
cell-permeable ceramide. J Biol Chem. 272:24154–24158. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vafeiadou K, Weech M, Sharma V, Yaqoob P,
Todd S, Williams CM, Jackson KG and Lovegrove JA: A review of the
evidence for the effects of total dietary fat, saturated,
monounsaturated and n-6 polyunsaturated fatty acids on vascular
function, endothelial progenitor cells and microparticles. Br J
Nutr. 107:303–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang CL, Lyngmo V and Nordøy A: The
effects of saturated fatty acids on endothelial cells. Thromb Res.
65:65–75. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Artwohl M, Roden M, Waldhäusl W,
Freudenthaler A and Baumgartner-Parzer SM: Free fatty acids trigger
apoptosis and inhibit cell cycle progression in human vascular
endothelial cells. FASEB J. 18:146–148. 2004.PubMed/NCBI
|
|
25
|
Yu H, Yang Z, Ding X, Wang Y and Han Y:
Correlation between the different chain lengths of free fatty acid
oxidation and ability of trophoblastic invasion. Chin Med J (Engl).
127:3378–3382. 2014.PubMed/NCBI
|
|
26
|
Dimmeler S, Haendeler J, Galle J and
Zeiher AM: Oxidized low-density lipoprotein induces apoptosis of
human endothelial cells by activation of CPP32-like proteases. A
mechanistic clue to the ‘response to injury’ hypothesis.
Circulation. 95:1760–1763. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sata M and Walsh K: Oxidized LDL activates
fas-mediated endothelial cell apoptosis. J Clin Invest.
102:1682–1689. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Raymond MA, Désormeaux A, Laplante P,
Vigneault N, Filep JG, Landry K, Pshezhetsky AV and Hébert MJ:
Apoptosis of endothelial cells triggers a caspase-dependent
anti-apoptotic paracrine loop active on VSMC. FASEB J. 18:705–707.
2004.PubMed/NCBI
|
|
29
|
Laplante P, Raymond MA, Gagnon G,
Vigneault N, Sasseville AM, Langelier Y, Bernard M, Raymond Y and
Hébert MJ: Novel fibrogenic pathways are activated in response to
endothelial apoptosis: Implications in the pathophysiology of
systemic sclerosis. J Immunol. 174:5740–5749. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li D, Yang B and Mehta JL: Ox-LDL induces
apoptosis in human coronary artery endothelial cells: Role of PKC,
PTK, bcl-2, and Fas. Am J Physiol. 275:H568–H576. 1998.PubMed/NCBI
|
|
31
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cirman T, Oresić K, Mazovec GD, Turk V,
Reed JC, Myers RM, Salvesen GS and Turk B: Selective disruption of
lysosomes in HeLa cells triggers apoptosis mediated by cleavage of
Bid by multiple papain-like lysosomal cathepsins. J Biol Chem.
279:3578–3587. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heinrich M, Neumeyer J, Jakob M, Hallas C,
Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W,
Trauzold A, Hethke A and Schütze S: Cathepsin D links TNF-induced
acid sphingomyelinase to Bid-mediated caspase-9 and −3 activation.
Cell Death Differ. 11:550–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stoka V, Turk B, Schendel SL, Kim TH,
Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson
M, et al: Lysosomal protease pathways to apoptosis. Cleavage of
bid, not pro-caspases, is the most likely route. J Biol Chem.
276:3149–3157. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guicciardi ME, Leist M and Gores GJ:
Lysosomes in cell death. Oncogene. 23:2881–2890. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schotte P, Van Criekinge W, Van de Craen
M, Van Loo G, Desmedt M, Grooten J, Cornelissen M, De Ridder L,
Vandekerckhove J, Fiers W, et al: Cathepsin B-mediated activation
of the proinflammatory caspase-11. Biochem Biophys Res Commun.
251:379–387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vancompernolle K, Van Herreweghe F,
Pynaert G, Van de Craen M, De Vos K, Totty N, Sterling A, Fiers W,
Vandenabeele P and Grooten J: Atractyloside-induced release of
cathepsin B, a protease with caspase-processing activity. FEBS
Lett. 438:150–158. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishisaka R, Utsumi T, Kanno T, Arita K,
Katunuma N, Akiyama J and Utsumi K: Participation of a cathepsin
L-type protease in the activation of caspase-3. Cell Struct Funct.
24:465–470. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ishisaka R, Kanno T, Akiyama J, Yoshioka
T, Utsumi K and Utsumi T: Activation of caspase-3 by lysosomal
cysteine proteases and its role in
2,2′-azobis-(2-amidinopropane)dihydrochloride (AAPH)-induced
apoptosis in HL-60 cells. J Biochem. 129:35–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsai JY, Lee MJ, Dah-Tsyr Chang M and
Huang H: The effect of catalase on migration and invasion of lung
cancer cells by regulating the activities of cathepsin S, L, and K.
Exp Cell Res. 323:28–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Q, Han M, Wang W, Song Y, Chen G,
Wang Z and Liang Z: Downregulation of cathepsin L suppresses cancer
invasion and migration by inhibiting transforming growth
factor-β-mediated epithelial-mesenchymal transition. Oncol Rep.
33:1851–1859. 2015.PubMed/NCBI
|
|
42
|
Burden RE, Gormley JA, Jaquin TJ, Small
DM, Quinn DJ, Hegarty SM, Ward C, Walker B, Johnston JA, Olwill SA
and Scott CJ: Antibody-mediated inhibition of cathepsin S blocks
colorectal tumor invasion and angiogenesis. Clin Cancer Res.
15:6042–6051. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li X, Cheng XW, Hu L, Wu H, Guo-Ping, Hao
CN, Jiang H, Zhu E, Huang Z, Inoue A, et al: Cathepsin S activity
controls ischemia-induced neovascularization in mice. Int J
Cardiol. 183:198–208. 2015. View Article : Google Scholar : PubMed/NCBI
|